当前位置:
X-MOL 学术
›
Nat. Struct. Mol. Biol.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
An ultra high-throughput method for single-cell joint analysis of open chromatin and transcriptome.
Nature Structural & Molecular Biology ( IF 16.8 ) Pub Date : 2019-11-06 , DOI: 10.1038/s41594-019-0323-x Chenxu Zhu 1 , Miao Yu 1 , Hui Huang 1, 2 , Ivan Juric 3 , Armen Abnousi 3 , Rong Hu 1 , Jacinta Lucero 4 , M Margarita Behrens 4 , Ming Hu 3 , Bing Ren 1, 5
Nature Structural & Molecular Biology ( IF 16.8 ) Pub Date : 2019-11-06 , DOI: 10.1038/s41594-019-0323-x Chenxu Zhu 1 , Miao Yu 1 , Hui Huang 1, 2 , Ivan Juric 3 , Armen Abnousi 3 , Rong Hu 1 , Jacinta Lucero 4 , M Margarita Behrens 4 , Ming Hu 3 , Bing Ren 1, 5
Affiliation
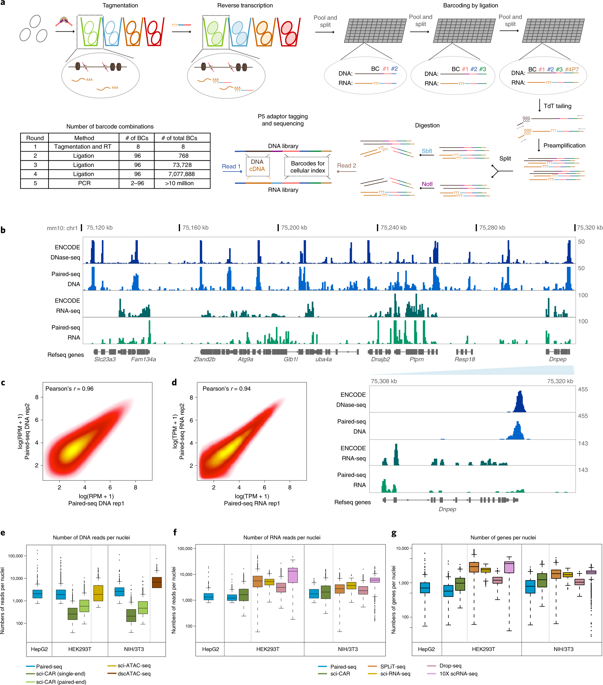
|
Simultaneous profiling of transcriptome and chromatin accessibility within single cells is a powerful approach to dissect gene regulatory programs in complex tissues. However, current tools are limited by modest throughput. We now describe an ultra high-throughput method, Paired-seq, for parallel analysis of transcriptome and accessible chromatin in millions of single cells. We demonstrate the utility of Paired-seq for analyzing the dynamic and cell-type-specific gene regulatory programs in complex tissues by applying it to mouse adult cerebral cortex and fetal forebrain. The joint profiles of a large number of single cells allowed us to deconvolute the transcriptome and open chromatin landscapes in the major cell types within these brain tissues, infer putative target genes of candidate enhancers, and reconstruct the trajectory of cellular lineages within the developing forebrain.
中文翻译:

开放染色质和转录组单细胞联合分析的超高通量方法。
同时分析单细胞内的转录组和染色质可及性是剖析复杂组织中基因调控程序的有力方法。然而,目前的工具受到适度吞吐量的限制。我们现在描述一种超高通量方法,Paired-seq,用于并行分析数百万个单细胞中的转录组和可接近的染色质。我们通过将 Paired-seq 应用于小鼠成人大脑皮层和胎儿前脑,展示了 Paired-seq 在分析复杂组织中动态和细胞类型特异性基因调控程序的效用。大量单细胞的联合特征使我们能够解卷积这些脑组织内主要细胞类型中的转录组和开放染色质景观,推断候选增强子的推定靶基因,
更新日期:2019-11-06
中文翻译:
开放染色质和转录组单细胞联合分析的超高通量方法。
同时分析单细胞内的转录组和染色质可及性是剖析复杂组织中基因调控程序的有力方法。然而,目前的工具受到适度吞吐量的限制。我们现在描述一种超高通量方法,Paired-seq,用于并行分析数百万个单细胞中的转录组和可接近的染色质。我们通过将 Paired-seq 应用于小鼠成人大脑皮层和胎儿前脑,展示了 Paired-seq 在分析复杂组织中动态和细胞类型特异性基因调控程序的效用。大量单细胞的联合特征使我们能够解卷积这些脑组织内主要细胞类型中的转录组和开放染色质景观,推断候选增强子的推定靶基因,


























 京公网安备 11010802027423号
京公网安备 11010802027423号