Experimental & Molecular Medicine ( IF 9.5 ) Pub Date : 2019-11-06 , DOI: 10.1038/s12276-019-0337-9 Eric Seidel 1, 2, 3 , Julia Schewe 1, 2, 3 , Ute I Scholl 1, 2, 3
|
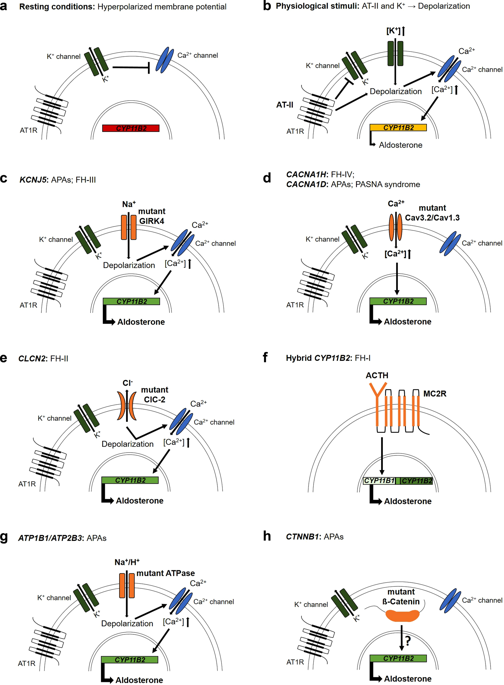
Primary aldosteronism is characterized by at least partially autonomous production of the adrenal steroid hormone aldosterone and is the most common cause of secondary hypertension. The most frequent subforms are idiopathic hyperaldosteronism and aldosterone-producing adenoma. Rare causes include unilateral hyperplasia, adrenocortical carcinoma and Mendelian forms (familial hyperaldosteronism). Studies conducted in the last eight years have identified somatic driver mutations in a substantial portion of aldosterone-producing adenomas, including the genes KCNJ5 (encoding inwardly rectifying potassium channel GIRK4), CACNA1D (encoding a subunit of L-type voltage-gated calcium channel CaV1.3), ATP1A1 (encoding a subunit of Na+/K+-ATPase), ATP2B3 (encoding a Ca2+-ATPase), and CTNNB1 (encoding ß-catenin). In addition, aldosterone-producing cells were recently reported to form small clusters (aldosterone-producing cell clusters) beneath the adrenal capsule. Such clusters accumulate with age and appear to be more frequent in individuals with idiopathic hyperaldosteronism. The fact that they are associated with somatic mutations implicated in aldosterone-producing adenomas also suggests a precursor function for adenomas. Rare germline variants of CYP11B2 (encoding aldosterone synthase), CLCN2 (encoding voltage-gated chloride channel ClC-2), KCNJ5, CACNA1H (encoding a subunit of T-type voltage-gated calcium channel CaV3.2), and CACNA1D have been reported in different subtypes of familial hyperaldosteronism. Collectively, these studies suggest that primary aldosteronism is largely due to genetic mutations in single genes, with potential implications for diagnosis and therapy.
中文翻译:
原发性醛固酮增多症的遗传原因。
原发性醛固酮增多症的特征是肾上腺类固醇激素醛固酮至少部分自主产生,是继发性高血压的最常见原因。最常见的亚型是特发性醛固酮增多症和产生醛固酮的腺瘤。罕见的原因包括单侧增生、肾上腺皮质癌和孟德尔形式(家族性醛固酮增多症)。在过去八年中进行的研究已经确定了大部分产生醛固酮的腺瘤中的体细胞驱动突变,包括基因KCNJ5(编码内向整流钾通道 GIRK4)、CACNA1D(编码 L 型电压门控钙通道 Ca 的亚基)V 1.3), ATP1A1 (编码 Na 的一个亚基+ /K + -ATPase)、ATP2B3(编码 Ca 2+ -ATPase)和CTNNB1(编码 ß-catenin)。此外,最近有报道称,产生醛固酮的细胞在肾上腺囊下方形成小簇(产生醛固酮的细胞簇)。这种簇随着年龄的增长而积累,并且似乎在患有特发性醛固酮增多症的个体中更常见。它们与与产生醛固酮的腺瘤有关的体细胞突变相关的事实也表明了腺瘤的前体功能。CYP11B2(编码醛固酮合酶)、CLCN2(编码电压门控氯离子通道 ClC-2)、KCNJ5、CACNA1H的罕见种系变体(编码 T 型电压门控钙通道 Ca V 3.2 的一个亚基)和CACNA1D在不同亚型的家族性醛固酮增多症中均有报道。总的来说,这些研究表明原发性醛固酮增多症主要是由于单个基因的基因突变,对诊断和治疗具有潜在意义。










































 京公网安备 11010802027423号
京公网安备 11010802027423号