当前位置:
X-MOL 学术
›
Surf. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Low-index surface energies, cleavage energies, and surface relaxations for crystalline NiAl from first-principles calculations
Surface Science ( IF 2.1 ) Pub Date : 2020-05-01 , DOI: 10.1016/j.susc.2019.121532 Linxia Wang , King C. Lai , Li Huang , James W. Evans , Yong Han
Surface Science ( IF 2.1 ) Pub Date : 2020-05-01 , DOI: 10.1016/j.susc.2019.121532 Linxia Wang , King C. Lai , Li Huang , James W. Evans , Yong Han
|
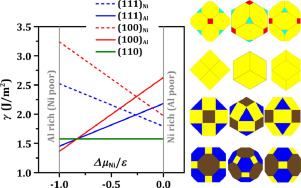
Abstract NiAl surfaces frequently serve as a platform for studying a broad range of physical and chemical phenomena including chemisorption, catalysis, oxidation, alloy growth, and surface nanostructure formation. Knowledge of precise values for low-index surface energies of NiAl, the most fundamental quantities characterizing surface thermodynamics, is often invaluable for understanding of these phenomena. In all previous analyses for NiAl(100) and NiAl(111), Ni- and Al-terminations are not distinguished, and half of the cleavage energy (or equivalently, the average of the surface energies of two differently terminated surfaces) is always identified as the “surface energy”. No values are available for individual surface energies of Ni- or Al-terminated NiAl(100) or NiAl(111) surfaces, whereas knowledge of only cleavage energy is often insufficient for analyzing surface-associated behavior. In this work, we perform extensive first-principles density-functional-theory (DFT) calculations for surface energies and cleavage energies of NiAl(110), NiAl(100) and NiAl(111) by considering the chemical-potential-based formulations to clarify the ambiguity in their surface energies and cleavage energies. We obtain a surface energy phase diagram for these three low-index surfaces versus the relevant chemical potential, as well as the chemical-potential-dependent Wulff plots for NiAl crystal equilibrium shapes. We also provide the surface-relaxation information from our DFT calculations for comparison with previous experimental data.
中文翻译:

从第一性原理计算得到的结晶 NiAl 的低指数表面能、解理能和表面弛豫
摘要 NiAl 表面经常用作研究广泛的物理和化学现象的平台,包括化学吸附、催化、氧化、合金生长和表面纳米结构的形成。了解 NiAl 低指数表面能的精确值(表征表面热力学的最基本量)对于理解这些现象通常是非常宝贵的。在之前对 NiAl(100) 和 NiAl(111) 的所有分析中,没有区分 Ni 和 Al 末端,并且总是确定一半的解理能(或等效地,两个不同末端表面的表面能的平均值)作为“表面能”。没有可用于 Ni 或 Al 封端的 NiAl(100) 或 NiAl(111) 表面的单个表面能的值,而仅了解解理能通常不足以分析表面相关行为。在这项工作中,我们通过考虑基于化学势的公式,对 NiAl(110)、NiAl(100) 和 NiAl(111) 的表面能和解理能进行了广泛的第一性原理密度泛函理论 (DFT) 计算澄清它们的表面能和解理能的模糊性。我们获得了这三个低指数表面与相关化学势的表面能相图,以及 NiAl 晶体平衡形状的化学势相关伍尔夫图。我们还提供了来自 DFT 计算的表面松弛信息,以便与以前的实验数据进行比较。我们通过考虑基于化学势的公式,对 NiAl(110)、NiAl(100) 和 NiAl(111) 的表面能和解理能进行广泛的第一性原理密度泛函理论 (DFT) 计算,以澄清在它们的表面能和解理能。我们获得了这三个低指数表面与相关化学势的表面能相图,以及 NiAl 晶体平衡形状的化学势相关伍尔夫图。我们还提供了来自 DFT 计算的表面松弛信息,以便与以前的实验数据进行比较。我们通过考虑基于化学势的公式,对 NiAl(110)、NiAl(100) 和 NiAl(111) 的表面能和解理能进行广泛的第一性原理密度泛函理论 (DFT) 计算,以澄清在它们的表面能和解理能。我们获得了这三个低指数表面与相关化学势的表面能相图,以及 NiAl 晶体平衡形状的化学势相关伍尔夫图。我们还提供了来自 DFT 计算的表面松弛信息,以便与以前的实验数据进行比较。NiAl(100) 和 NiAl(111) 通过考虑基于化学势的公式来澄清其表面能和解理能的模糊性。我们获得了这三个低指数表面与相关化学势的表面能相图,以及 NiAl 晶体平衡形状的化学势相关伍尔夫图。我们还提供了来自 DFT 计算的表面松弛信息,以便与以前的实验数据进行比较。NiAl(100) 和 NiAl(111) 通过考虑基于化学势的公式来澄清其表面能和解理能的模糊性。我们获得了这三个低指数表面与相关化学势的表面能相图,以及 NiAl 晶体平衡形状的化学势相关伍尔夫图。我们还提供了来自 DFT 计算的表面松弛信息,以便与以前的实验数据进行比较。
更新日期:2020-05-01
中文翻译:
从第一性原理计算得到的结晶 NiAl 的低指数表面能、解理能和表面弛豫
摘要 NiAl 表面经常用作研究广泛的物理和化学现象的平台,包括化学吸附、催化、氧化、合金生长和表面纳米结构的形成。了解 NiAl 低指数表面能的精确值(表征表面热力学的最基本量)对于理解这些现象通常是非常宝贵的。在之前对 NiAl(100) 和 NiAl(111) 的所有分析中,没有区分 Ni 和 Al 末端,并且总是确定一半的解理能(或等效地,两个不同末端表面的表面能的平均值)作为“表面能”。没有可用于 Ni 或 Al 封端的 NiAl(100) 或 NiAl(111) 表面的单个表面能的值,而仅了解解理能通常不足以分析表面相关行为。在这项工作中,我们通过考虑基于化学势的公式,对 NiAl(110)、NiAl(100) 和 NiAl(111) 的表面能和解理能进行了广泛的第一性原理密度泛函理论 (DFT) 计算澄清它们的表面能和解理能的模糊性。我们获得了这三个低指数表面与相关化学势的表面能相图,以及 NiAl 晶体平衡形状的化学势相关伍尔夫图。我们还提供了来自 DFT 计算的表面松弛信息,以便与以前的实验数据进行比较。我们通过考虑基于化学势的公式,对 NiAl(110)、NiAl(100) 和 NiAl(111) 的表面能和解理能进行广泛的第一性原理密度泛函理论 (DFT) 计算,以澄清在它们的表面能和解理能。我们获得了这三个低指数表面与相关化学势的表面能相图,以及 NiAl 晶体平衡形状的化学势相关伍尔夫图。我们还提供了来自 DFT 计算的表面松弛信息,以便与以前的实验数据进行比较。我们通过考虑基于化学势的公式,对 NiAl(110)、NiAl(100) 和 NiAl(111) 的表面能和解理能进行广泛的第一性原理密度泛函理论 (DFT) 计算,以澄清在它们的表面能和解理能。我们获得了这三个低指数表面与相关化学势的表面能相图,以及 NiAl 晶体平衡形状的化学势相关伍尔夫图。我们还提供了来自 DFT 计算的表面松弛信息,以便与以前的实验数据进行比较。NiAl(100) 和 NiAl(111) 通过考虑基于化学势的公式来澄清其表面能和解理能的模糊性。我们获得了这三个低指数表面与相关化学势的表面能相图,以及 NiAl 晶体平衡形状的化学势相关伍尔夫图。我们还提供了来自 DFT 计算的表面松弛信息,以便与以前的实验数据进行比较。NiAl(100) 和 NiAl(111) 通过考虑基于化学势的公式来澄清其表面能和解理能的模糊性。我们获得了这三个低指数表面与相关化学势的表面能相图,以及 NiAl 晶体平衡形状的化学势相关伍尔夫图。我们还提供了来自 DFT 计算的表面松弛信息,以便与以前的实验数据进行比较。










































 京公网安备 11010802027423号
京公网安备 11010802027423号