npj Computational Materials ( IF 9.4 ) Pub Date : 2019-10-25 , DOI: 10.1038/s41524-019-0240-x Tibor Szilvási , Benjamin W. J. Chen , Manos Mavrikakis
|
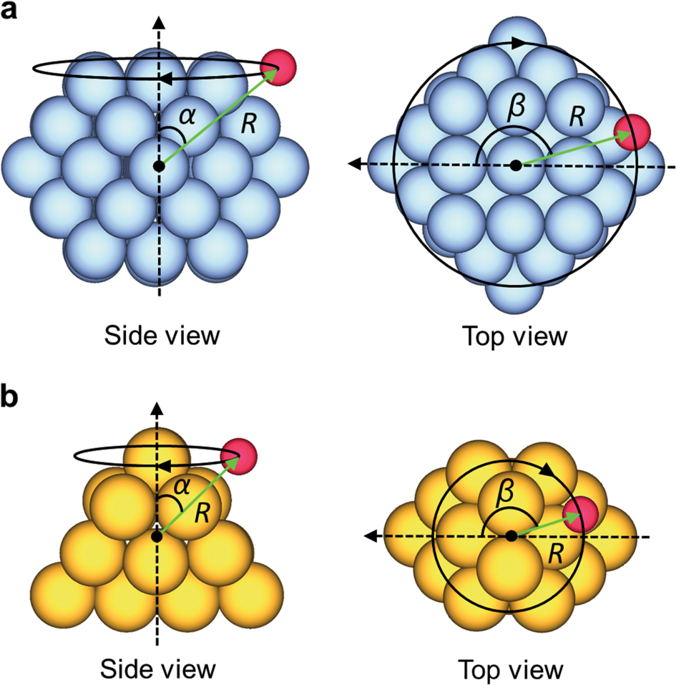
The diverse coordination environments on the surfaces of discrete, three-dimensional (3D) nanoclusters contribute significantly to their unique catalytic properties. Identifying the numerous adsorption sites and diffusion paths on these clusters is however tedious and time-consuming, especially for large, asymmetric nanoclusters. Here, we present a simple, automated method for constructing approximate 2D potential energy surfaces for the adsorption of atomic species on the surfaces of 3D nanoclusters with minimal human intervention. These potential energy surfaces fully characterize the important adsorption sites and diffusion paths on the nanocluster surfaces with accuracies similar to current approaches and at comparable computational cost. Our method can treat complex nanoclusters, such as alloy nanoclusters, and accounts for cluster relaxation and adsorbate-induced reconstruction, important for obtaining accurate energetics. Moreover, its highly parallelizable nature is ideal for modern supercomputer architectures. We showcase our method using two clusters: Au18 and Pt55. For Au18, diffusion of atomic hydrogen between the most stable sites occurs via non-intuitive paths, underlining the necessity of exploring the complete potential energy surface. By enabling the rapid and unbiased assessment of adsorption and diffusion on large, complex nanoclusters, which are particularly difficult to handle manually, our method will help advance materials discovery and the rational design of catalysts.
中文翻译:
识别纳米团簇表面上稳定的吸附位点和扩散路径:自动扫描算法
离散的三维(3D)纳米团簇表面上的多种配位环境对其独特的催化性能做出了重要贡献。然而,鉴定这些簇上的大量吸附位点和扩散路径是繁琐且耗时的,尤其是对于大型,不对称的纳米团簇而言。在这里,我们提出了一种简单的自动化方法,可在无需人工干预的情况下,构建近似的2D势能表面来吸附3D纳米团簇表面上的原子种类。这些势能表面以与当前方法相似的精确度和可比较的计算成本,完全表征了纳米簇表面上的重要吸附位点和扩散路径。我们的方法可以处理复杂的纳米团簇,例如合金纳米团簇,并解释了团簇松弛和吸附物诱导的重建,这对于获得准确的能量学很重要。此外,其高度可并行化的特性非常适合现代超级计算机体系结构。我们使用两个聚类来展示我们的方法:Au18和Pt 55。对于Au 18,原子氢在最稳定位点之间的扩散通过非直觉路径发生,这强调了探索完整势能面的必要性。通过对大型,复杂的纳米团簇(尤其是手动处理)的吸附和扩散进行快速而公正的评估,我们的方法将有助于推进材料发现和催化剂的合理设计。








































 京公网安备 11010802027423号
京公网安备 11010802027423号