当前位置:
X-MOL 学术
›
Surf. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Hydrogen Adsorption on Ir(111), Ir(100) and Ir(110) – Surface and Coverage Dependence
Surface Science ( IF 2.1 ) Pub Date : 2020-02-01 , DOI: 10.1016/j.susc.2019.121514 Chunli Liu , Ling Zhu , Xiaodong Wen , Yong Yang , Yong-Wang Li , Haijun Jiao
Surface Science ( IF 2.1 ) Pub Date : 2020-02-01 , DOI: 10.1016/j.susc.2019.121514 Chunli Liu , Ling Zhu , Xiaodong Wen , Yong Yang , Yong-Wang Li , Haijun Jiao
|
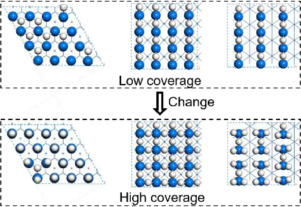
Abstract Hydrogen adsorption on the perfect Ir(111) as well as the metastable and unreconstructed Ir(100) and Ir(110) surfaces up to saturation coverage has been systematically computed using periodic density functional theory and ab initio atomistic thermodynamics for understanding the interaction mechanism of hydrogen on iridium surfaces. On the Ir(111) surface including van der Waals dispersion, hydrogen adsorption prefers the threefold hollow sites at low coverage and the top sites at high coverage; in agreement with the experiments (Phys. Rev. B 60 (1999) 14016). The computed adsorption energy and desorption temperature of hydrogen agree with the experiments [−0.57 (fcc-3H) and −0.53 (hcp-3H) vs. −0.55 eV; 180 and 325 K vs. 190 and 310 K, respectively]. On the Ir (100) surface, the bridge adsorption sites are preferred in the whole coverage range, in agreement with the LEED pattern (Phys. Rev. B 73 (2006) 75430), however, adsorption energy and desorption temperature are slightly overestimated by including van der Waals dispersion (−1.50 vs. −1.02 ± 0.15 eV; 470 vs. 425 – 389 K). On the Ir(110) surface, short-bridge sites are preferred at low coverage and the top sites become dominant at high coverage, and the calculated desorption temperatures are close to experiments by including van der Waals dispersion (210 and 365 K vs. 220 and 375 K). At low coverage, the different configurations of hydrogen adsorption on the Ir(111) have the similar energies, indicating their negligible repulsive interaction, while the Ir(100) and Ir(110) surfaces prefer regular line-shape adsorption configurations due to attractive interaction, and such adsorption configurations have not been observed experimentally. Our results show that differences in adsorption configurations and energies are associated with their differences in surface structures, and in turn explain the need of different methods in computing the adsorption properties on different surfaces. Such surface-dependent properties should also be possible on other metal surfaces.
中文翻译:

Ir(111)、Ir(100) 和 Ir(110) 上的氢吸附 - 表面和覆盖率相关性
摘要 使用周期密度泛函理论和 ab initio 原子热力学了解相互作用机制,系统地计算了完美 Ir(111) 以及亚稳态和未重构 Ir(100) 和 Ir(110) 表面直至饱和覆盖的氢吸附铱表面上的氢。在包括范德华分散的 Ir(111) 表面上,氢吸附优先选择低覆盖率的三重空心位置和高覆盖率的顶部位置;与实验一致(Phys. Rev. B 60 (1999) 14016)。计算出的氢气吸附能和解吸温度与实验结果一致 [-0.57 (fcc-3H) 和 -0.53 (hcp-3H) vs. -0.55 eV; 180 和 325 K 与 190 和 310 K,分别]。在 Ir (100) 表面,与 LEED 模式 (Phys. Rev. B 73 (2006) 75430) 一致,在整个覆盖范围内优选桥吸附位点,但是,通过包括范德华分散 (-1.50) 稍微高估了吸附能和解吸温度对比 -1.02 ± 0.15 eV;470 对比 425 – 389 K)。在 Ir(110) 表面,短桥位点在低覆盖率下优先,顶部位点在高覆盖率下占主导地位,计算的解吸温度接近实验,包括范德华色散(210 和 365 K vs. 220和 375 K)。在低覆盖率下,Ir(111) 上不同的氢吸附构型具有相似的能量,表明它们的排斥相互作用可以忽略不计,而 Ir(100) 和 Ir(110) 表面由于吸引力相互作用而更喜欢规则的线形吸附构型, 并且这种吸附配置尚未在实验中观察到。我们的结果表明,吸附构型和能量的差异与其表面结构的差异有关,进而解释了计算不同表面吸附特性的不同方法的必要性。这种依赖于表面的特性也应该适用于其他金属表面。
更新日期:2020-02-01
中文翻译:
Ir(111)、Ir(100) 和 Ir(110) 上的氢吸附 - 表面和覆盖率相关性
摘要 使用周期密度泛函理论和 ab initio 原子热力学了解相互作用机制,系统地计算了完美 Ir(111) 以及亚稳态和未重构 Ir(100) 和 Ir(110) 表面直至饱和覆盖的氢吸附铱表面上的氢。在包括范德华分散的 Ir(111) 表面上,氢吸附优先选择低覆盖率的三重空心位置和高覆盖率的顶部位置;与实验一致(Phys. Rev. B 60 (1999) 14016)。计算出的氢气吸附能和解吸温度与实验结果一致 [-0.57 (fcc-3H) 和 -0.53 (hcp-3H) vs. -0.55 eV; 180 和 325 K 与 190 和 310 K,分别]。在 Ir (100) 表面,与 LEED 模式 (Phys. Rev. B 73 (2006) 75430) 一致,在整个覆盖范围内优选桥吸附位点,但是,通过包括范德华分散 (-1.50) 稍微高估了吸附能和解吸温度对比 -1.02 ± 0.15 eV;470 对比 425 – 389 K)。在 Ir(110) 表面,短桥位点在低覆盖率下优先,顶部位点在高覆盖率下占主导地位,计算的解吸温度接近实验,包括范德华色散(210 和 365 K vs. 220和 375 K)。在低覆盖率下,Ir(111) 上不同的氢吸附构型具有相似的能量,表明它们的排斥相互作用可以忽略不计,而 Ir(100) 和 Ir(110) 表面由于吸引力相互作用而更喜欢规则的线形吸附构型, 并且这种吸附配置尚未在实验中观察到。我们的结果表明,吸附构型和能量的差异与其表面结构的差异有关,进而解释了计算不同表面吸附特性的不同方法的必要性。这种依赖于表面的特性也应该适用于其他金属表面。










































 京公网安备 11010802027423号
京公网安备 11010802027423号