当前位置:
X-MOL 学术
›
ACS Earth Space Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Ab Initio Molecular Dynamics Simulation of Divalent Metal Cation Incorporation in Calcite: Implications for Interpreting X-ray Absorption Spectroscopy Data
ACS Earth and Space Chemistry ( IF 2.9 ) Pub Date : 2019-11-01 , DOI: 10.1021/acsearthspacechem.9b00247 Sebastien N. Kerisit 1 , Micah P. Prange 1
ACS Earth and Space Chemistry ( IF 2.9 ) Pub Date : 2019-11-01 , DOI: 10.1021/acsearthspacechem.9b00247 Sebastien N. Kerisit 1 , Micah P. Prange 1
Affiliation
|
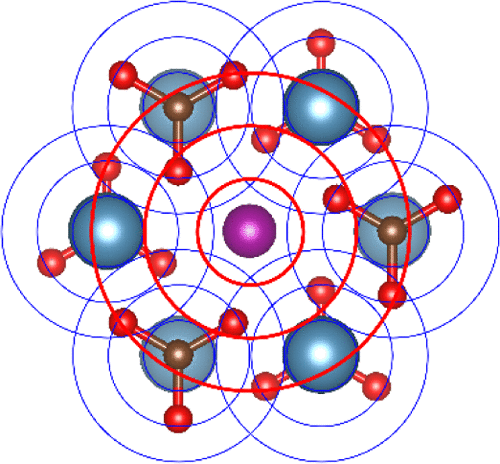
Calcite (CaCO3) is a ubiquitous mineral with the ability to accommodate a wide range of impurities. Determination of the coordination environment and incorporation modes of impurities in calcite has historically relied primarily on the interpretation of extended X-ray absorption fine structure (EXAFS) spectroscopy. However, lack of standards combined with the large number of degrees of freedom involved in shell-by-shell fits have made the interpretation of EXAFS spectra challenging. In this work, ab initio molecular dynamics (AIMD) simulations were performed to investigate the incorporation of seven divalent metal cation impurities, namely, Ba2+, Pb2+, Sr2+, Cd2+, Mn2+, Co2+, and Zn2+, in calcite. These cations span a wide range of sizes: 30.7 to −22.8% change in ionic radius with respect to Ca2+. The atomic trajectories were then used to compute EXAFS spectra for direct comparison with published experimental spectra. The simulations confirmed that all seven metal cations incorporate in calcite via substitution in the calcium site. The AIMD–EXAFS approach allowed for overcoming limitations of the shell-by-shell fitting approach, such as the difficulties in resolving weak backscatterers beyond the first coordination shell. As a result, the AIMD–EXAFS approach was able to provide a detailed and comprehensive characterization of the structural relaxation around divalent metal cation impurities in calcite.
中文翻译:

方解石中二价金属阳离子结合的从头算分子动力学模拟:解释X射线吸收光谱数据的意义
方解石(CaCO 3)是一种普遍存在的矿物,能够容纳各种杂质。历史上,方解石中配位环境和杂质掺入方式的确定主要依赖于扩展X射线吸收精细结构(EXAFS)光谱学的解释。但是,缺乏标准以及逐壳拟合所涉及的大量自由度使对EXAFS光谱的解释具有挑战性。在这项工作中,进行了从头算分子动力学(AIMD)模拟以研究7种二价金属阳离子杂质的掺入,即Ba 2 +,Pb 2 +,Sr 2 +,Cd 2 +,Mn 2 +,Co方解石中的2+和Zn 2+。这些阳离子的尺寸范围很广:相对于Ca 2+,离子半径的变化为30.7至-22.8%。然后将原子轨迹用于计算EXAFS光谱,以便直接与已发布的实验光谱进行比较。模拟证实,所有七个金属阳离子都通过在钙位点的取代而掺入方解石中。AIMD-EXAFS方法克服了逐壳拟合方法的局限性,例如难以解决第一个协调壳之外的弱后向散射问题。结果,AIMD-EXAFS方法能够对方解石中二价金属阳离子杂质周围的结构弛豫进行详细而全面的表征。
更新日期:2019-11-01
中文翻译:
方解石中二价金属阳离子结合的从头算分子动力学模拟:解释X射线吸收光谱数据的意义
方解石(CaCO 3)是一种普遍存在的矿物,能够容纳各种杂质。历史上,方解石中配位环境和杂质掺入方式的确定主要依赖于扩展X射线吸收精细结构(EXAFS)光谱学的解释。但是,缺乏标准以及逐壳拟合所涉及的大量自由度使对EXAFS光谱的解释具有挑战性。在这项工作中,进行了从头算分子动力学(AIMD)模拟以研究7种二价金属阳离子杂质的掺入,即Ba 2 +,Pb 2 +,Sr 2 +,Cd 2 +,Mn 2 +,Co方解石中的2+和Zn 2+。这些阳离子的尺寸范围很广:相对于Ca 2+,离子半径的变化为30.7至-22.8%。然后将原子轨迹用于计算EXAFS光谱,以便直接与已发布的实验光谱进行比较。模拟证实,所有七个金属阳离子都通过在钙位点的取代而掺入方解石中。AIMD-EXAFS方法克服了逐壳拟合方法的局限性,例如难以解决第一个协调壳之外的弱后向散射问题。结果,AIMD-EXAFS方法能够对方解石中二价金属阳离子杂质周围的结构弛豫进行详细而全面的表征。










































 京公网安备 11010802027423号
京公网安备 11010802027423号