npj Computational Materials ( IF 9.4 ) Pub Date : 2019-10-11 , DOI: 10.1038/s41524-019-0236-6 Noam Bernstein , Gábor Csányi , Volker L. Deringer
|
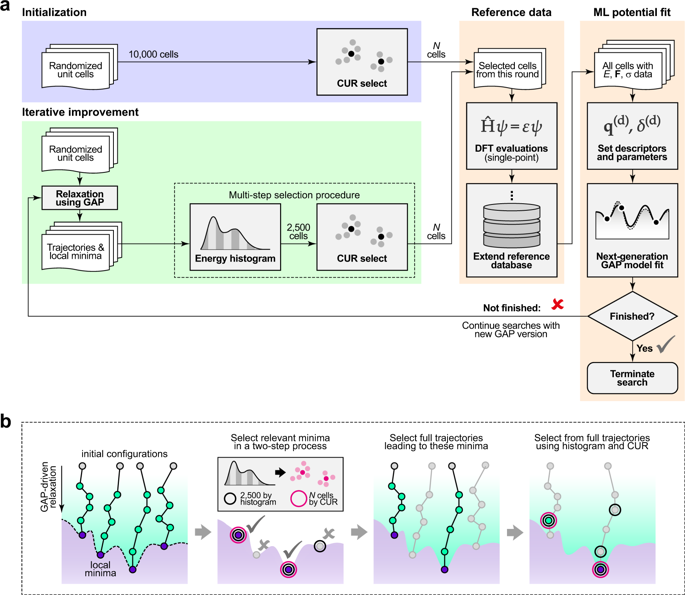
Interatomic potential models based on machine learning (ML) are rapidly developing as tools for material simulations. However, because of their flexibility, they require large fitting databases that are normally created with substantial manual selection and tuning of reference configurations. Here, we show that ML potentials can be built in a largely automated fashion, exploring and fitting potential-energy surfaces from the beginning (de novo) within one and the same protocol. The key enabling step is the use of a configuration-averaged kernel metric that allows one to select the few most relevant and diverse structures at each step. The resulting potentials are accurate and robust for the wide range of configurations that occur during structure searching, despite only requiring a relatively small number of single-point DFT calculations on small unit cells. We apply the method to materials with diverse chemical nature and coordination environments, marking an important step toward the more routine application of ML potentials in physics, chemistry, and materials science.
中文翻译:
从头开始探索和自导学习势能面
基于机器学习(ML)的原子间势模型正在迅速发展为材料模拟的工具。但是,由于它们的灵活性,它们需要大型的拟合数据库,这些数据库通常是通过大量的手动选择和参考配置调整来创建的。在这里,我们证明了可以以一种高度自动化的方式建立ML势能,并从一开始就在一个相同的协议中探索并拟合势能面。关键的使能步骤是使用配置平均的内核度量,该度量允许每个步骤选择一些最相关且多样化的结构。对于在结构搜索过程中发生的各种配置,所产生的电势是准确而稳健的,尽管只需要在较小的单位单元上进行相对少量的单点DFT计算即可。我们将该方法应用于具有多种化学性质和配位环境的材料,这标志着ML势在物理,化学和材料科学中更常规的应用迈出了重要的一步。








































 京公网安备 11010802027423号
京公网安备 11010802027423号