当前位置:
X-MOL 学术
›
Photochem. Photobiol. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Theoretical insight into the photophysical properties of six heteroleptic Ir(iii) phosphorescent complexes bearing ppy-type ligands†
Photochemical & Photobiological Sciences ( IF 2.7 ) Pub Date : 2019-10-01 , DOI: 10.1039/c9pp00218a Deming Han 1, 2, 3, 4, 5 , Lihui Zhao 1, 2, 3, 4 , Xuerong Han 1, 2, 3, 4
Photochemical & Photobiological Sciences ( IF 2.7 ) Pub Date : 2019-10-01 , DOI: 10.1039/c9pp00218a Deming Han 1, 2, 3, 4, 5 , Lihui Zhao 1, 2, 3, 4 , Xuerong Han 1, 2, 3, 4
Affiliation
|
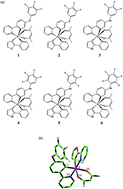
By using density functional theory and time-dependent density functional theory, the geometrical, electronic and photophysical properties of six complexes with two ppy-type ligands and one acetylacetone anion around the Ir center have been explored. The lowest energy absorption wavelengths are located at 414 nm for 1, 434 nm for 2, 434 nm for 3, 421 nm for 4, 436 nm for 5, and 425 nm for 6, respectively. The lowest energy emissions of these complexes are localized at 617, 492, 633, 634, 491 and 491 nm, respectively, for complexes 1–6, simulated in CH2Cl2 medium at the M062X level. The calculated lowest lying absorption wavelength and the lowest energy emission wavelength for complex 3 are very close to the available experimental values. The position and number of the incorporated electron-withdrawing fluorine substituents have some effect on the electronic and photophysical properties of these studied complexes.
中文翻译:

对六种带有ppy型配体的Ir(iii)磷杂光配合物的光物理性质的理论洞察力†
通过使用密度泛函理论和随时间变化的密度泛函理论,研究了在Ir中心周围具有两个ppy型配体和一个乙酰丙酮阴离子的六种配合物的几何,电子和光物理性质。最低的能量吸收波长分别位于414 nm(对于1),434 nm(对于2),434(3),421 nm(对于4),436 nm(对于5)和425 nm(对于6)。在CH 2 Cl 2中模拟的复合物1–6,这些复合物的最低能量发射分别位于617、492、633、634、491和491 nm。M062X级别的中级。计算出的配合物3的最低吸收波长和最低能量发射波长非常接近可用的实验值。引入的吸电子氟取代基的位置和数目对这些研究的配合物的电子和光物理性质有一定影响。
更新日期:2019-11-06
中文翻译:
对六种带有ppy型配体的Ir(iii)磷杂光配合物的光物理性质的理论洞察力†
通过使用密度泛函理论和随时间变化的密度泛函理论,研究了在Ir中心周围具有两个ppy型配体和一个乙酰丙酮阴离子的六种配合物的几何,电子和光物理性质。最低的能量吸收波长分别位于414 nm(对于1),434 nm(对于2),434(3),421 nm(对于4),436 nm(对于5)和425 nm(对于6)。在CH 2 Cl 2中模拟的复合物1–6,这些复合物的最低能量发射分别位于617、492、633、634、491和491 nm。M062X级别的中级。计算出的配合物3的最低吸收波长和最低能量发射波长非常接近可用的实验值。引入的吸电子氟取代基的位置和数目对这些研究的配合物的电子和光物理性质有一定影响。










































 京公网安备 11010802027423号
京公网安备 11010802027423号