Nature Chemistry ( IF 19.2 ) Pub Date : 2019-09-30 , DOI: 10.1038/s41557-019-0337-3 Zhendong Li 1 , Sheng Guo 1 , Qiming Sun 1 , Garnet Kin-Lic Chan 1
|
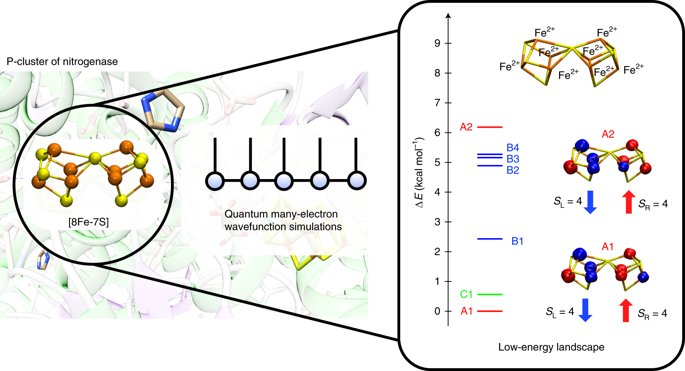
The electronic structure of the nitrogenase metal cofactors is central to nitrogen fixation. However, the P-cluster and FeMo cofactor, each containing eight Fe atoms, have eluded detailed characterization of their electronic properties. We report on the low-energy electronic states of the P-cluster in three oxidation states through exhaustive many-electron wavefunction simulations enabled by new theoretical methods. The energy scales of orbital and spin excitations overlap, yielding a dense spectrum with features that we trace to the underlying atomic states and recouplings. The clusters exist in superpositions of spin configurations with non-classical spin correlations, complicating interpretation of magnetic spectroscopies, whereas the charges are mostly localized from reorganization of the cluster and its surroundings. On oxidation, the opening of the P-cluster substantially increases the density of states, which is intriguing given its proposed role in electron transfer. These results demonstrate that many-electron simulations stand to provide new insights into the electronic structure of the nitrogenase cofactors.
中文翻译:
通过多电子量子波函数模拟显示的氮酶P簇的电子态势。
固氮酶金属辅因子的电子结构对固氮至关重要。但是,每个都包含八个Fe原子的P簇和FeMo辅助因子,尚未对其电子性质进行详细的表征。我们通过新的理论方法实现的详尽的多电子波函数模拟,报告了在三个氧化态下P团簇的低能电子态。轨道和自旋激发的能级重叠,产生了一个密集光谱,其特征可以追溯到基本的原子态和耦合。这些团簇以自旋构型的叠加形式存在,具有非经典的自旋相关性,从而使磁谱解释变得复杂,而电荷大多集中在该团簇及其周围环境的重组中。氧化时 P团簇的打开大大增加了态密度,考虑到它在电子转移中的作用,这很有趣。这些结果表明,多电子模拟有望为了解固氮酶辅助因子的电子结构提供新的见解。










































 京公网安备 11010802027423号
京公网安备 11010802027423号