当前位置:
X-MOL 学术
›
Mol. Syst. Des. Eng.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Identification of optimally stable nanocluster geometries via mathematical optimization and density-functional theory
Molecular Systems Design & Engineering ( IF 3.2 ) Pub Date : 2019/09/25 , DOI: 10.1039/c9me00108e Natalie M. Isenberg 1, 2, 3, 4 , Michael G. Taylor 3, 4, 5, 6 , Zihao Yan 3, 4, 5, 6 , Christopher L. Hanselman 1, 2, 3, 4 , Giannis Mpourmpakis 3, 4, 5, 6 , Chrysanthos E. Gounaris 1, 2, 3, 4
Molecular Systems Design & Engineering ( IF 3.2 ) Pub Date : 2019/09/25 , DOI: 10.1039/c9me00108e Natalie M. Isenberg 1, 2, 3, 4 , Michael G. Taylor 3, 4, 5, 6 , Zihao Yan 3, 4, 5, 6 , Christopher L. Hanselman 1, 2, 3, 4 , Giannis Mpourmpakis 3, 4, 5, 6 , Chrysanthos E. Gounaris 1, 2, 3, 4
Affiliation
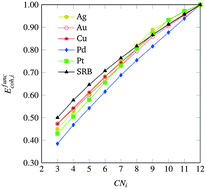
|
Small nanoparticles, a.k.a. nanoclusters, of transition metals have been studied extensively for a wide range of applications due to their highly tunable properties dependent on size, structure, and composition. For these small particles, there has been considerable effort towards theoretically predicting what is the most energetically favorable arrangement of atoms when forming a nanocluster. In this work, we develop a computational framework that couples density-functional theory calculations with mathematical optimization modeling to identify highly stable, mono-metallic transition metal nanoclusters of various sizes. This is accomplished by devising and solving a rigorous mathematical optimization model that maximizes a general cohesive energy function to obtain nanocluster structures of provably maximal cohesiveness. We then utilize density-functional theory calculations and error term regression to identify model corrections that are necessary to account with better accuracy for different transition metals. This allows us to encode metal-specific, analytical functions for cohesive energy into a mathematical optimization-based framework that can accurately predict which nanocluster geometries will be most cohesive according to density-functional theory calculations. We employ our framework in the context of Ag, Au, Cu, Pd and Pt, and we present sequences of highly cohesive nanoclusters for sizes up to 100 atoms, yielding insights into structures that might be experimentally accessible and/or structures that could be used as model nanoclusters for further study.
中文翻译:

通过数学优化和密度泛函理论确定最佳稳定的纳米簇几何形状
由于过渡金属的大小,结构和组成的高度可调性,它们被广泛地用于各种应用的过渡金属中,因此被广泛研究。对于这些小颗粒,在理论上预测形成纳米团簇时最有力的原子排列方面已经付出了巨大的努力。在这项工作中,我们开发了一个计算框架,该框架将密度泛函理论计算与数学优化模型相结合,以识别各种尺寸的高度稳定的单金属过渡金属纳米簇。这是通过设计和求解严格的数学优化模型来实现的,该模型使一般的内聚能函数最大化,从而获得具有可证明的最大内聚力的纳米簇结构。然后,我们利用密度泛函理论计算和误差项回归来确定模型校正,这些校正对于更好地计算不同过渡金属的准确性是必要的。这使我们可以将金属的内聚能分析函数编码到基于数学优化的框架中,该框架可以根据密度泛函理论计算准确预测哪些纳米簇几何形状内聚性最高。我们在Ag,Au,Cu,Pd和Pt的背景下采用了我们的框架,并提出了具有高度凝聚力的纳米簇的序列,这些簇的大小最大为100个原子,从而洞察了可以通过实验获得的结构和/或可以使用的结构作为模型纳米簇有待进一步研究。
更新日期:2020-02-13
中文翻译:
通过数学优化和密度泛函理论确定最佳稳定的纳米簇几何形状
由于过渡金属的大小,结构和组成的高度可调性,它们被广泛地用于各种应用的过渡金属中,因此被广泛研究。对于这些小颗粒,在理论上预测形成纳米团簇时最有力的原子排列方面已经付出了巨大的努力。在这项工作中,我们开发了一个计算框架,该框架将密度泛函理论计算与数学优化模型相结合,以识别各种尺寸的高度稳定的单金属过渡金属纳米簇。这是通过设计和求解严格的数学优化模型来实现的,该模型使一般的内聚能函数最大化,从而获得具有可证明的最大内聚力的纳米簇结构。然后,我们利用密度泛函理论计算和误差项回归来确定模型校正,这些校正对于更好地计算不同过渡金属的准确性是必要的。这使我们可以将金属的内聚能分析函数编码到基于数学优化的框架中,该框架可以根据密度泛函理论计算准确预测哪些纳米簇几何形状内聚性最高。我们在Ag,Au,Cu,Pd和Pt的背景下采用了我们的框架,并提出了具有高度凝聚力的纳米簇的序列,这些簇的大小最大为100个原子,从而洞察了可以通过实验获得的结构和/或可以使用的结构作为模型纳米簇有待进一步研究。










































 京公网安备 11010802027423号
京公网安备 11010802027423号