当前位置:
X-MOL 学术
›
Faraday Discuss.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Direct dynamics analysis of the cationic Cp*(PMe3)Ir(CH3) methane C–H activation mechanism
Faraday Discussions ( IF 3.3 ) Pub Date : 2019-09-16 , DOI: 10.1039/c9fd00035f Ryan Carlsen 1, 2, 3, 4 , Jordan R. Jenkins 1, 2, 3, 4 , Daniel H. Ess 1, 2, 3, 4
Faraday Discussions ( IF 3.3 ) Pub Date : 2019-09-16 , DOI: 10.1039/c9fd00035f Ryan Carlsen 1, 2, 3, 4 , Jordan R. Jenkins 1, 2, 3, 4 , Daniel H. Ess 1, 2, 3, 4
Affiliation
|
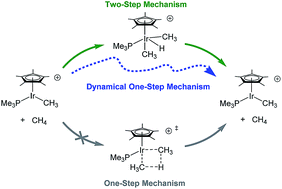
For the σ-bond metathesis reaction between methane and cationic Cp*(PMe3)IrIII(CH3), previous static DFT calculations revealed a two-step oxidative addition/reductive elimination mechanism with an intervening IrV–H intermediate. We recently reported quasiclassical direct molecular dynamics simulations where starting from the vibrationally-averaged oxidative addition transition state a minor, but significant, amount of trajectories bypassed the IrV–H intermediate in a dynamical one-step mechanism. These trajectories also revealed that after the reductive coupling transition state is passed methane always dissociates and the C–H σ-complex is skipped. Here we report direct dynamics simulations using a microcanonical temperature sampling method with trajectories initialized and propagated using the Gaussian program. Sets of 30 trajectories were examined with average time steps ranging from 1.5 to 0.25 femtoseconds (fs). These results showed that a step size of ∼1.5 fs is too large and likely overestimates the amount of trajectories skipping the IrV–H intermediate. Trajectories with 0.75 and 0.25 fs time steps gave qualitatively similar profiles compared to our previous results using our program DynSuite with a 1 fs time step. Reverse trajectories with an average time step of 0.25 fs showed complete skipping of the methane σ complex. 30 trajectories were also propagated using a SMD continuum dichloromethane solvent model. These trajectories showed very similar behavior to gas-phase trajectories.
中文翻译:

阳离子Cp *(PMe 3)Ir(CH 3)甲烷C–H活化机理的 直接动力学分析
对于甲烷和阳离子Cp *(PMe 3)Ir III(CH 3)之间的σ键易位反应,先前的静态DFT计算显示了一个中间有Ir V –H中间体的两步氧化加成/还原消除机理。我们最近报道了准经典的直接分子动力学模拟,其中从振动平均的氧化加成过渡态开始,少量但重要的轨迹绕过Ir V–H在动态一步机制中的中间体。这些轨迹还表明,在通过还原偶联跃迁状态后,甲烷始终会解离,而C–Hσ络合物会被跳过。在这里,我们报告使用微规范温度采样方法的直接动力学模拟,并使用高斯程序对轨迹进行初始化和传播。检查了30条轨迹的集合,其平均时间步长为1.5到0.25飞秒(fs)。这些结果表明,约1.5 fs的步长太大,可能会高估跳过Ir V的轨迹数量-H中间体。0.75和0.25 fs时间步长的轨迹与我们以前使用1 fs时间步长的程序DynSuite的结果相比,在质量上具有相似的轮廓。反向轨迹的平均时间步长为0.25 fs,表明甲烷σ络合物完全跳过。使用SMD连续介质二氯甲烷溶剂模型还传播了30条轨迹。这些轨迹显示出与气相轨迹非常相似的行为。
更新日期:2019-12-04
中文翻译:
阳离子Cp *(PMe 3)Ir(CH 3)甲烷C–H活化机理的 直接动力学分析
对于甲烷和阳离子Cp *(PMe 3)Ir III(CH 3)之间的σ键易位反应,先前的静态DFT计算显示了一个中间有Ir V –H中间体的两步氧化加成/还原消除机理。我们最近报道了准经典的直接分子动力学模拟,其中从振动平均的氧化加成过渡态开始,少量但重要的轨迹绕过Ir V–H在动态一步机制中的中间体。这些轨迹还表明,在通过还原偶联跃迁状态后,甲烷始终会解离,而C–Hσ络合物会被跳过。在这里,我们报告使用微规范温度采样方法的直接动力学模拟,并使用高斯程序对轨迹进行初始化和传播。检查了30条轨迹的集合,其平均时间步长为1.5到0.25飞秒(fs)。这些结果表明,约1.5 fs的步长太大,可能会高估跳过Ir V的轨迹数量-H中间体。0.75和0.25 fs时间步长的轨迹与我们以前使用1 fs时间步长的程序DynSuite的结果相比,在质量上具有相似的轮廓。反向轨迹的平均时间步长为0.25 fs,表明甲烷σ络合物完全跳过。使用SMD连续介质二氯甲烷溶剂模型还传播了30条轨迹。这些轨迹显示出与气相轨迹非常相似的行为。










































 京公网安备 11010802027423号
京公网安备 11010802027423号