当前位置:
X-MOL 学术
›
Chem. Res. Toxicol.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Quantitative Predictions for Molecular Initiating Events Using Three-Dimensional Quantitative Structure-Activity Relationships.
Chemical Research in Toxicology ( IF 3.7 ) Pub Date : 2019-12-17 , DOI: 10.1021/acs.chemrestox.9b00136 Timothy E H Allen 1 , Jonathan M Goodman 1 , Steve Gutsell 2 , Paul J Russell 2
Chemical Research in Toxicology ( IF 3.7 ) Pub Date : 2019-12-17 , DOI: 10.1021/acs.chemrestox.9b00136 Timothy E H Allen 1 , Jonathan M Goodman 1 , Steve Gutsell 2 , Paul J Russell 2
Affiliation
|
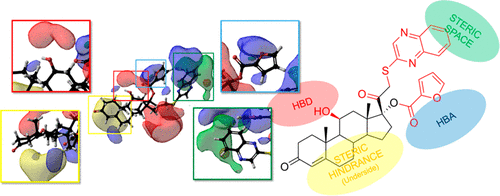
The aim of human toxicity risk assessment is to determine a safe dose or exposure to a chemical for humans. This requires an understanding of the exposure of a person to a chemical and how much of the chemical is required to cause an adverse effect. To do this computationally, we need to understand how much of a chemical is required to perturb normal biological function in an adverse outcome pathway (AOP). The molecular initiating event (MIE) is the first step in an adverse outcome pathway and can be considered as a chemical interaction between a chemical toxicant and a biological molecule. Key chemical characteristics can be identified and used to model the chemistry of these MIEs. In this study, we do just this by using chemical substructures to categorize chemicals and 3D quantitative structure-activity relationships (QSARs) based on comparative molecular field analysis (CoMFA) to calculate molecular activity. Models have been constructed across a variety of human biological targets, the glucocorticoid receptor, mu opioid receptor, cyclooxygenase-2 enzyme, human ether-à-go-go related gene channel, and dopamine transporter. These models tend to provide molecular activity estimation well within one log unit and electronic and steric fields that can be visualized to better understand the MIE and biological target of interest. The outputs of these fields can be used to identify key aspects of a chemical's chemistry which can be changed to reduce its ability to activate a given MIE. With this methodology, the quantitative chemical activity can be predicted for a wide variety of MIEs, which can feed into AOP-based chemical risk assessments, and understanding of the chemistry behind the MIE can be gained.
中文翻译:

使用三维定量结构-活性关系的分子引发事件的定量预测。
人体毒性风险评估的目的是确定人类的安全剂量或化学药品的暴露量。这需要了解一个人接触某种化学药品以及需要多少化学药品才能产生不利影响。为此,我们需要了解在不良结局途径(AOP)中需要多少化学物质才能扰乱正常的生物学功能。分子引发事件(MIE)是不良后果途径中的第一步,可以视为化学毒物与生物分子之间的化学相互作用。可以识别关键化学特征并将其用于模拟这些MIE的化学性质。在这项研究中,我们通过使用化学亚结构对化学物质进行分类,并基于比较分子场分析(CoMFA)来计算分子活性,从而实现3D定量构效关系(QSAR)。已经跨多种人类生物学靶标构建了模型,这些靶标包括糖皮质激素受体,μ阿片受体,环加氧酶2酶,与人类醚相关的基因通道和多巴胺转运蛋白。这些模型趋向于在一个对数单位内以及分子和电子场中提供良好的分子活性估计,可以可视化以更好地了解MIE和感兴趣的生物学靶标。这些字段的输出可用于识别化学物质化学的关键方面,可以对其进行更改以降低其激活给定MIE的能力。通过这种方法,
更新日期:2019-12-18
中文翻译:
使用三维定量结构-活性关系的分子引发事件的定量预测。
人体毒性风险评估的目的是确定人类的安全剂量或化学药品的暴露量。这需要了解一个人接触某种化学药品以及需要多少化学药品才能产生不利影响。为此,我们需要了解在不良结局途径(AOP)中需要多少化学物质才能扰乱正常的生物学功能。分子引发事件(MIE)是不良后果途径中的第一步,可以视为化学毒物与生物分子之间的化学相互作用。可以识别关键化学特征并将其用于模拟这些MIE的化学性质。在这项研究中,我们通过使用化学亚结构对化学物质进行分类,并基于比较分子场分析(CoMFA)来计算分子活性,从而实现3D定量构效关系(QSAR)。已经跨多种人类生物学靶标构建了模型,这些靶标包括糖皮质激素受体,μ阿片受体,环加氧酶2酶,与人类醚相关的基因通道和多巴胺转运蛋白。这些模型趋向于在一个对数单位内以及分子和电子场中提供良好的分子活性估计,可以可视化以更好地了解MIE和感兴趣的生物学靶标。这些字段的输出可用于识别化学物质化学的关键方面,可以对其进行更改以降低其激活给定MIE的能力。通过这种方法,






































 京公网安备 11010802027423号
京公网安备 11010802027423号