Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Integrative Protein Modeling in RosettaNMR from Sparse Paramagnetic Restraints.
Structure ( IF 5.7 ) Pub Date : 2019-09-12 , DOI: 10.1016/j.str.2019.08.012 Georg Kuenze 1 , Richard Bonneau 2 , Julia Koehler Leman 3 , Jens Meiler 1
Structure ( IF 5.7 ) Pub Date : 2019-09-12 , DOI: 10.1016/j.str.2019.08.012 Georg Kuenze 1 , Richard Bonneau 2 , Julia Koehler Leman 3 , Jens Meiler 1
Affiliation
|
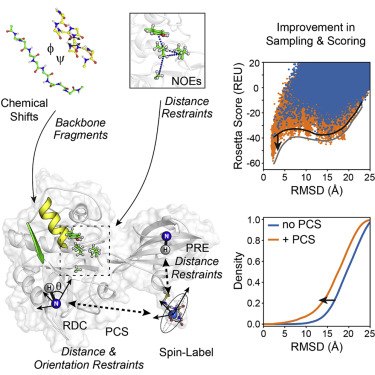
Computational methods to predict protein structure from nuclear magnetic resonance (NMR) restraints that only require assignment of backbone signals, hold great potential to study larger proteins. Ideally, computational methods designed to work with sparse data need to add atomic detail that is missing in the experimental restraints. We introduce a comprehensive framework into the Rosetta suite that uses NMR restraints derived from paramagnetic labeling. Specifically, RosettaNMR incorporates pseudocontact shifts, residual dipolar couplings, and paramagnetic relaxation enhancements. It continues to use backbone chemical shifts and nuclear Overhauser effect distance restraints. We assess RosettaNMR for protein structure prediction by folding 28 monomeric proteins and 8 homo-oligomeric proteins. Furthermore, the general applicability of RosettaNMR is demonstrated on two protein-protein and three protein-ligand docking examples. Paramagnetic restraints generated more accurate models for 85% of the benchmark proteins and, when combined with chemical shifts, sampled high-accuracy models (≤2Å) in 50% of the cases.
中文翻译:

来自稀疏顺磁约束的 RosettaNMR 中的综合蛋白质建模。
从核磁共振 (NMR) 限制预测蛋白质结构的计算方法只需要分配主干信号,具有研究更大蛋白质的巨大潜力。理想情况下,设计用于处理稀疏数据的计算方法需要添加实验约束中缺少的原子细节。我们在 Rosetta 套件中引入了一个综合框架,该框架使用源自顺磁标记的 NMR 约束。具体来说,RosettaNMR 结合了赝接触位移、残余偶极耦合和顺磁弛豫增强。它继续使用主干化学位移和核 Overhauser 效应距离限制。我们通过折叠 28 个单体蛋白和 8 个同源寡聚蛋白来评估 RosettaNMR 的蛋白质结构预测。此外,RosettaNMR 的普遍适用性在两个蛋白质-蛋白质和三个蛋白质-配体对接示例中得到证明。顺磁约束为 85% 的基准蛋白质生成了更准确的模型,并且当与化学位移相结合时,在 50% 的情况下采样了高精度模型 (≤2Å)。
更新日期:2019-09-12
中文翻译:
来自稀疏顺磁约束的 RosettaNMR 中的综合蛋白质建模。
从核磁共振 (NMR) 限制预测蛋白质结构的计算方法只需要分配主干信号,具有研究更大蛋白质的巨大潜力。理想情况下,设计用于处理稀疏数据的计算方法需要添加实验约束中缺少的原子细节。我们在 Rosetta 套件中引入了一个综合框架,该框架使用源自顺磁标记的 NMR 约束。具体来说,RosettaNMR 结合了赝接触位移、残余偶极耦合和顺磁弛豫增强。它继续使用主干化学位移和核 Overhauser 效应距离限制。我们通过折叠 28 个单体蛋白和 8 个同源寡聚蛋白来评估 RosettaNMR 的蛋白质结构预测。此外,RosettaNMR 的普遍适用性在两个蛋白质-蛋白质和三个蛋白质-配体对接示例中得到证明。顺磁约束为 85% 的基准蛋白质生成了更准确的模型,并且当与化学位移相结合时,在 50% 的情况下采样了高精度模型 (≤2Å)。


























 京公网安备 11010802027423号
京公网安备 11010802027423号