当前位置:
X-MOL 学术
›
WIREs Comput. Mol. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Classical molecular dynamics on graphics processing unit architectures
Wiley Interdisciplinary Reviews: Computational Molecular Science ( IF 16.8 ) Pub Date : 2019-09-05 , DOI: 10.1002/wcms.1444 Ádám Jász 1 , Ádám Rák 1 , István Ladjánszki 1 , György Cserey 2
Wiley Interdisciplinary Reviews: Computational Molecular Science ( IF 16.8 ) Pub Date : 2019-09-05 , DOI: 10.1002/wcms.1444 Ádám Jász 1 , Ádám Rák 1 , István Ladjánszki 1 , György Cserey 2
Affiliation
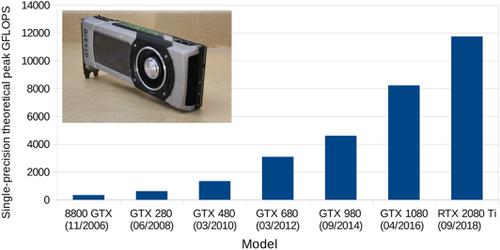
|
Molecular dynamics (MD) has experienced a significant growth in the recent decades. Simulating systems consisting of hundreds of thousands of atoms is a routine task of computational chemistry researchers nowadays. Thanks to the straightforwardly parallelizable structure of the algorithms, the most promising method to speed‐up MD calculations is exploiting the large‐scale processing power offered by the parallel hardware architecture of graphics processing units or GPUs. Programming GPUs is becoming easier with general‐purpose GPU computing frameworks and higher levels of abstraction. In the recent years, implementing MD simulations on graphics processors has gained a large interest, with multiple popular software packages including some form of GPU‐acceleration support. Different approaches have been developed regarding various aspects of the algorithms, with important differences in the specific solutions. Focusing on published works in the field of classical MD, we describe the chosen implementation methods and algorithmic techniques used for porting to GPU, as well as how recent advances of GPU architectures will provide even more optimization possibilities in the future.
中文翻译:

图形处理单元体系结构上的经典分子动力学
在最近的几十年中,分子动力学(MD)经历了显着增长。如今,由数十万个原子组成的模拟系统是计算化学研究人员的一项日常任务。由于算法的直接可并行化结构,加快MD计算的最有前途的方法是利用图形处理单元或GPU的并行硬件体系结构提供的大规模处理能力。通用GPU计算框架和更高级别的抽象,使GPU编程变得越来越容易。近年来,在图形处理器上实现MD仿真已经引起了人们的极大兴趣,它提供了多种流行的软件包,其中包括某种形式的GPU加速支持。已经针对算法的各个方面开发了不同的方法,但在具体解决方案中存在重要差异。以经典MD领域中已发表的作品为重点,我们描述了用于移植到GPU的所选实现方法和算法技术,以及GPU架构的最新进展将如何在将来提供更多的优化可能性。
更新日期:2019-09-05
中文翻译:
图形处理单元体系结构上的经典分子动力学
在最近的几十年中,分子动力学(MD)经历了显着增长。如今,由数十万个原子组成的模拟系统是计算化学研究人员的一项日常任务。由于算法的直接可并行化结构,加快MD计算的最有前途的方法是利用图形处理单元或GPU的并行硬件体系结构提供的大规模处理能力。通用GPU计算框架和更高级别的抽象,使GPU编程变得越来越容易。近年来,在图形处理器上实现MD仿真已经引起了人们的极大兴趣,它提供了多种流行的软件包,其中包括某种形式的GPU加速支持。已经针对算法的各个方面开发了不同的方法,但在具体解决方案中存在重要差异。以经典MD领域中已发表的作品为重点,我们描述了用于移植到GPU的所选实现方法和算法技术,以及GPU架构的最新进展将如何在将来提供更多的优化可能性。










































 京公网安备 11010802027423号
京公网安备 11010802027423号