当前位置:
X-MOL 学术
›
npj Syst. Biol. Appl.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
iOmicsPASS: network-based integration of multiomics data for predictive subnetwork discovery.
npj Systems Biology and Applications ( IF 3.5 ) Pub Date : 2019-07-09 , DOI: 10.1038/s41540-019-0099-y Hiromi W L Koh 1, 2 , Damian Fermin 3 , Christine Vogel 4 , Kwok Pui Choi 5 , Rob M Ewing 6 , Hyungwon Choi 1, 2, 7
npj Systems Biology and Applications ( IF 3.5 ) Pub Date : 2019-07-09 , DOI: 10.1038/s41540-019-0099-y Hiromi W L Koh 1, 2 , Damian Fermin 3 , Christine Vogel 4 , Kwok Pui Choi 5 , Rob M Ewing 6 , Hyungwon Choi 1, 2, 7
Affiliation
|
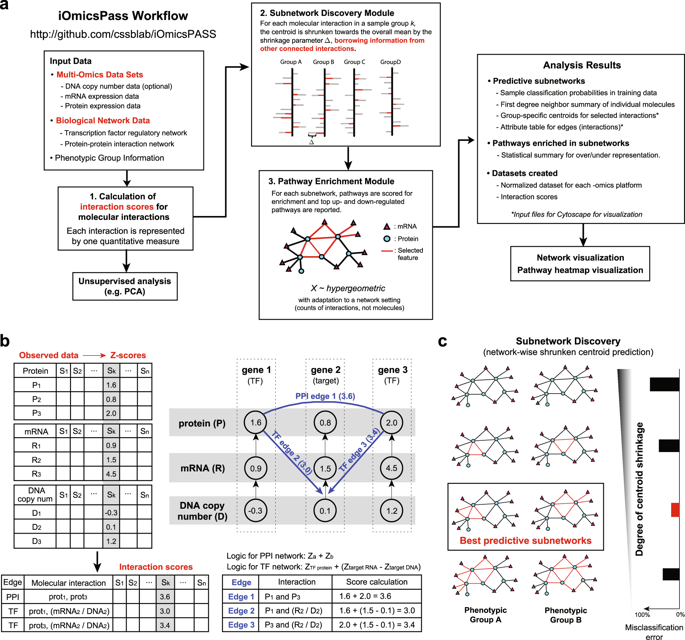
Computational tools for multiomics data integration have usually been designed for unsupervised detection of multiomics features explaining large phenotypic variations. To achieve this, some approaches extract latent signals in heterogeneous data sets from a joint statistical error model, while others use biological networks to propagate differential expression signals and find consensus signatures. However, few approaches directly consider molecular interaction as a data feature, the essential linker between different omics data sets. The increasing availability of genome-scale interactome data connecting different molecular levels motivates a new class of methods to extract interactive signals from multiomics data. Here we developed iOmicsPASS, a tool to search for predictive subnetworks consisting of molecular interactions within and between related omics data types in a supervised analysis setting. Based on user-provided network data and relevant omics data sets, iOmicsPASS computes a score for each molecular interaction, and applies a modified nearest shrunken centroid algorithm to the scores to select densely connected subnetworks that can accurately predict each phenotypic group. iOmicsPASS detects a sparse set of predictive molecular interactions without loss of prediction accuracy compared to alternative methods, and the selected network signature immediately provides mechanistic interpretation of the multiomics profile representing each sample group. Extensive simulation studies demonstrate clear benefit of interaction-level modeling. iOmicsPASS analysis of TCGA/CPTAC breast cancer data also highlights new transcriptional regulatory network underlying the basal-like subtype as positive protein markers, a result not seen through analysis of individual omics data.
中文翻译:

iOmicsPASS:基于网络的多组学数据集成,用于预测子网络发现。
用于多组学数据集成的计算工具通常被设计用于无监督地检测解释大表型变异的多组学特征。为了实现这一目标,一些方法从联合统计误差模型中提取异构数据集中的潜在信号,而另一些方法则使用生物网络来传播差异表达信号并找到共识签名。然而,很少有方法直接将分子相互作用视为数据特征,即不同组学数据集之间的重要链接。连接不同分子水平的基因组规模相互作用组数据的可用性不断增加,激发了一种从多组学数据中提取相互作用信号的新型方法。在这里,我们开发了 iOmicsPASS,这是一种在监督分析环境中搜索预测子网络的工具,该子网络由相关组学数据类型内部和之间的分子相互作用组成。根据用户提供的网络数据和相关组学数据集,iOmicsPASS 计算每个分子相互作用的分数,并对分数应用改进的最近收缩质心算法,以选择可以准确预测每个表型组的密集连接的子网络。与其他方法相比,iOmicsPASS 可以检测一组稀疏的预测分子相互作用,而不会损失预测准确性,并且所选的网络签名立即提供代表每个样本组的多组学概况的机械解释。广泛的模拟研究证明了交互级建模的明显好处。 iOmicsPASS 对 TCGA/CPTAC 乳腺癌数据的分析还强调了基底样亚型作为阳性蛋白标记物背后的新转录调控网络,这是通过单个组学数据分析未发现的结果。
更新日期:2019-07-09
中文翻译:
iOmicsPASS:基于网络的多组学数据集成,用于预测子网络发现。
用于多组学数据集成的计算工具通常被设计用于无监督地检测解释大表型变异的多组学特征。为了实现这一目标,一些方法从联合统计误差模型中提取异构数据集中的潜在信号,而另一些方法则使用生物网络来传播差异表达信号并找到共识签名。然而,很少有方法直接将分子相互作用视为数据特征,即不同组学数据集之间的重要链接。连接不同分子水平的基因组规模相互作用组数据的可用性不断增加,激发了一种从多组学数据中提取相互作用信号的新型方法。在这里,我们开发了 iOmicsPASS,这是一种在监督分析环境中搜索预测子网络的工具,该子网络由相关组学数据类型内部和之间的分子相互作用组成。根据用户提供的网络数据和相关组学数据集,iOmicsPASS 计算每个分子相互作用的分数,并对分数应用改进的最近收缩质心算法,以选择可以准确预测每个表型组的密集连接的子网络。与其他方法相比,iOmicsPASS 可以检测一组稀疏的预测分子相互作用,而不会损失预测准确性,并且所选的网络签名立即提供代表每个样本组的多组学概况的机械解释。广泛的模拟研究证明了交互级建模的明显好处。 iOmicsPASS 对 TCGA/CPTAC 乳腺癌数据的分析还强调了基底样亚型作为阳性蛋白标记物背后的新转录调控网络,这是通过单个组学数据分析未发现的结果。










































 京公网安备 11010802027423号
京公网安备 11010802027423号