Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Structure is more robust than other clustering methods in simulated mixed-ploidy populations
Heredity ( IF 3.1 ) Pub Date : 2019-07-08 , DOI: 10.1038/s41437-019-0247-6 Marc Stift 1 , Filip Kolář 2, 3 , Patrick G Meirmans 4
Heredity ( IF 3.1 ) Pub Date : 2019-07-08 , DOI: 10.1038/s41437-019-0247-6 Marc Stift 1 , Filip Kolář 2, 3 , Patrick G Meirmans 4
Affiliation
|
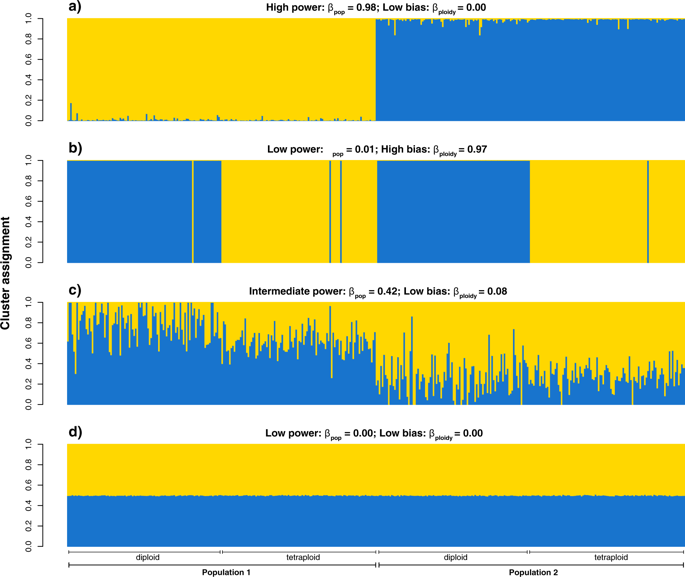
Analysis of population genetic structure has become a standard approach in population genetics. In polyploid complexes, clustering analyses can elucidate the origin of polyploid populations and patterns of admixture between different cytotypes. However, combining diploid and polyploid data can theoretically lead to biased inference with (artefactual) clustering by ploidy. We used simulated mixed-ploidy (diploid-autotetraploid) data to systematically compare the performance of k-means clustering and the model-based clustering methods implemented in Structure, Admixture, FastStructure and InStruct under different scenarios of differentiation and with different marker types. Under scenarios of strong population differentiation, the tested applications performed equally well. However, when population differentiation was weak, Structure was the only method that allowed unbiased inference with markers with limited genotypic information (co-dominant markers with unknown dosage or dominant markers). Still, since Structure was comparatively slow, the much faster but less powerful FastStructure provides a reasonable alternative for large datasets. Finally, although bias makes k-means clustering unsuitable for markers with incomplete genotype information, for large numbers of loci (>1000) with known dosage k-means clustering was superior to FastStructure in terms of power and speed. We conclude that Structure is the most robust method for the analysis of genetic structure in mixed-ploidy populations, although alternative methods should be considered under some specific conditions.
中文翻译:

在模拟混合倍性种群中,结构比其他聚类方法更稳健
群体遗传结构分析已成为群体遗传学的标准方法。在多倍体复合物中,聚类分析可以阐明多倍体种群的起源和不同细胞型之间的混合模式。然而,结合二倍体和多倍体数据理论上可能会导致通过倍性进行(人工)聚类的有偏见的推断。我们使用模拟混合倍体(二倍体-同源四倍体)数据系统地比较了 k-means 聚类的性能以及在不同分化场景和不同标记类型下在 Structure、Admixture、FastStructure 和 InStruct 中实施的基于模型的聚类方法的性能。在人口差异很大的情况下,测试的应用程序表现同样出色。然而,当人口分化较弱时,结构是唯一允许对具有有限基因型信息的标记(具有未知剂量的共显性标记或显性标记)进行无偏推断的方法。尽管如此,由于 Structure 相对较慢,因此速度更快但功能较弱的 FastStructure 为大型数据集提供了合理的替代方案。最后,尽管偏差使 k-means 聚类不适用于基因型信息不完整的标记,但对于具有已知剂量的大量基因座 (>1000),k-means 聚类在功率和速度方面优于 FastStructure。我们得出结论,结构是分析混合倍性种群遗传结构的最可靠方法,尽管在某些特定条件下应考虑替代方法。
更新日期:2019-07-08
中文翻译:
在模拟混合倍性种群中,结构比其他聚类方法更稳健
群体遗传结构分析已成为群体遗传学的标准方法。在多倍体复合物中,聚类分析可以阐明多倍体种群的起源和不同细胞型之间的混合模式。然而,结合二倍体和多倍体数据理论上可能会导致通过倍性进行(人工)聚类的有偏见的推断。我们使用模拟混合倍体(二倍体-同源四倍体)数据系统地比较了 k-means 聚类的性能以及在不同分化场景和不同标记类型下在 Structure、Admixture、FastStructure 和 InStruct 中实施的基于模型的聚类方法的性能。在人口差异很大的情况下,测试的应用程序表现同样出色。然而,当人口分化较弱时,结构是唯一允许对具有有限基因型信息的标记(具有未知剂量的共显性标记或显性标记)进行无偏推断的方法。尽管如此,由于 Structure 相对较慢,因此速度更快但功能较弱的 FastStructure 为大型数据集提供了合理的替代方案。最后,尽管偏差使 k-means 聚类不适用于基因型信息不完整的标记,但对于具有已知剂量的大量基因座 (>1000),k-means 聚类在功率和速度方面优于 FastStructure。我们得出结论,结构是分析混合倍性种群遗传结构的最可靠方法,尽管在某些特定条件下应考虑替代方法。








































 京公网安备 11010802027423号
京公网安备 11010802027423号