当前位置:
X-MOL 学术
›
Nat. Protoc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Systems structural biology measurements by in vivo cross-linking with mass spectrometry.
Nature Protocols ( IF 13.1 ) Pub Date : 2019-07-03 , DOI: 10.1038/s41596-019-0181-3 Juan D Chavez 1 , Jared P Mohr 1 , Martin Mathay 1 , Xuefei Zhong 1 , Andrew Keller 1 , James E Bruce 1
Nature Protocols ( IF 13.1 ) Pub Date : 2019-07-03 , DOI: 10.1038/s41596-019-0181-3 Juan D Chavez 1 , Jared P Mohr 1 , Martin Mathay 1 , Xuefei Zhong 1 , Andrew Keller 1 , James E Bruce 1
Affiliation
|
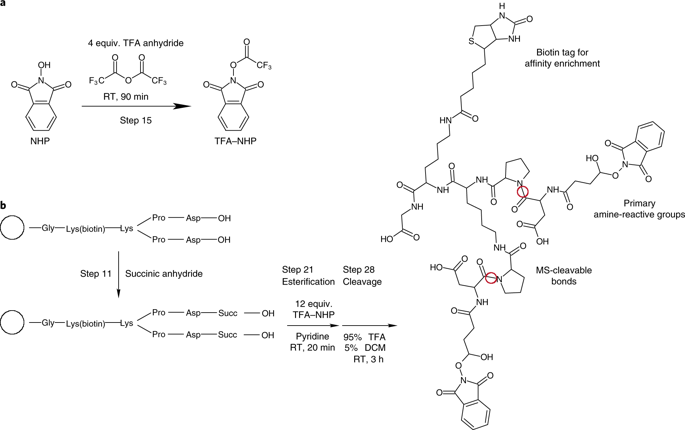
This protocol describes a workflow for utilizing large-scale cross-linking with mass spectrometry (XL-MS) to make systems-level structural biology measurements in complex biological samples, including cells, isolated organelles, and tissue samples. XL-MS is a structural biology technique that provides information on the molecular structure of proteins and protein complexes using chemical probes that report the proximity of probe-reactive amino acids within proteins, typically lysine residues. Information gained through XL-MS studies is often complementary to more traditional methods, such as X-ray crystallography, nuclear magnetic resonance, and cryo-electron microscopy. The use of MS-cleavable cross-linkers, including protein interaction reporter (PIR) technologies, enables XL-MS studies on protein structures and interactions in extremely complex biological samples, including intact living cells. PIR cross-linkers are designed to contain chemical bonds at specific locations within the cross-linker molecule that can be selectively cleaved by collision-induced dissociation or UV light. When broken, these bonds release the intact peptides that were cross-linked, as well as a reporter ion. Conservation of mass dictates that the sum of the two released peptide masses and the reporter mass equals the measured precursor mass. This relationship is used to identify cross-linked peptide pairs. Release of the individual peptides permits accurate measurement of their masses and independent amino acid sequence determination by tandem MS, allowing the use of standard proteomics search engines such as Comet for peptide sequence assignment, greatly simplifying data analysis of cross-linked peptide pairs. Search results are processed with XLinkProphet for validation and can be uploaded into XlinkDB for interaction network and structural analysis.
中文翻译:

通过体内交联质谱法进行系统结构生物学测量。
该协议描述了利用大规模交联与质谱 (XL-MS) 对复杂生物样品(包括细胞、分离的细胞器和组织样品)进行系统级结构生物学测量的工作流程。XL-MS 是一种结构生物学技术,它使用化学探针提供有关蛋白质和蛋白质复合物分子结构的信息,该探针报告蛋白质内与探针反应的氨基酸(通常是赖氨酸残基)的接近程度。通过 XL-MS 研究获得的信息通常是对更传统方法的补充,例如 X 射线晶体学、核磁共振和冷冻电子显微镜。使用 MS 可裂解交联剂,包括蛋白质相互作用报告基因 (PIR) 技术,能够对极其复杂的生物样品(包括完整的活细胞)中的蛋白质结构和相互作用进行 XL-MS 研究。PIR 交联剂被设计为在交联剂分子内的特定位置包含化学键,可以通过碰撞诱导解离或紫外光选择性裂解。当断裂时,这些键会释放交联的完整肽以及报告离子。质量守恒表明两个释放的肽质量和报告分子质量的总和等于测量的前体质量。这种关系用于识别交联的肽对。单个肽的释放允许通过串联质谱准确测量它们的质量和独立的氨基酸序列测定,允许使用标准蛋白质组学搜索引擎(如 Comet)进行肽序列分配,大大简化了交联肽对的数据分析。搜索结果经过 XLinkProphet 处理进行验证,并可上传至 XlinkDB 进行交互网络和结构分析。
更新日期:2019-11-18
中文翻译:
通过体内交联质谱法进行系统结构生物学测量。
该协议描述了利用大规模交联与质谱 (XL-MS) 对复杂生物样品(包括细胞、分离的细胞器和组织样品)进行系统级结构生物学测量的工作流程。XL-MS 是一种结构生物学技术,它使用化学探针提供有关蛋白质和蛋白质复合物分子结构的信息,该探针报告蛋白质内与探针反应的氨基酸(通常是赖氨酸残基)的接近程度。通过 XL-MS 研究获得的信息通常是对更传统方法的补充,例如 X 射线晶体学、核磁共振和冷冻电子显微镜。使用 MS 可裂解交联剂,包括蛋白质相互作用报告基因 (PIR) 技术,能够对极其复杂的生物样品(包括完整的活细胞)中的蛋白质结构和相互作用进行 XL-MS 研究。PIR 交联剂被设计为在交联剂分子内的特定位置包含化学键,可以通过碰撞诱导解离或紫外光选择性裂解。当断裂时,这些键会释放交联的完整肽以及报告离子。质量守恒表明两个释放的肽质量和报告分子质量的总和等于测量的前体质量。这种关系用于识别交联的肽对。单个肽的释放允许通过串联质谱准确测量它们的质量和独立的氨基酸序列测定,允许使用标准蛋白质组学搜索引擎(如 Comet)进行肽序列分配,大大简化了交联肽对的数据分析。搜索结果经过 XLinkProphet 处理进行验证,并可上传至 XlinkDB 进行交互网络和结构分析。










































 京公网安备 11010802027423号
京公网安备 11010802027423号