npj Materials Degradation ( IF 5.1 ) Pub Date : 2019-06-21 , DOI: 10.1038/s41529-019-0088-z Liang-Feng Huang , James M. Rondinelli
|
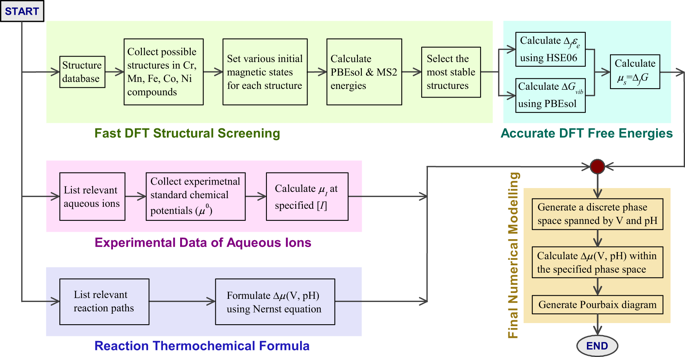
Magnetic transition metals (mTM = Cr, Mn, Fe, Co, and Ni) and their complex compounds (oxides, hydroxides, and oxyhydroxides) are highly important material platforms for diverse technologies, where electrochemical phase diagrams with respect to electrode potential and solution pH can be used to effectively understand their corrosion and oxidation behaviors in relevant aqueous environments. Many previous decades-old mTM–Pourbaix diagrams are inconsistent with various direct electrochemical observations, because experimental complexities associated with extracting reliable free energies of formation (ΔfG) lead to inaccuracies in the data used for modeling. Here, we develop a high-throughput simulation approach based on density-functional theory (DFT), which quickly screens structures and compounds using efficient DFT methods and calculates accurate ΔfG values, using high-level exchange-correlation functions to obtain ab initio Pourbaix diagrams in comprehensive and close agreement with various important electrochemical, geological, and biomagnetic observations reported over the last few decades. We also analyze the microscopic mechanisms governing the chemical trends among the ΔfG values and Pourbaix diagrams to further understand the electrochemical behaviors of mTM-based materials. Last, we provide probability profiles at variable electrode potential and solution pH to show quantitatively the likely coexistence of multiple-phase areas and diffuse phase boundaries.
中文翻译:
高通量从头算计算得出的磁性过渡金属及相关化合物的可靠电化学相图
磁性过渡金属(mTM = Cr,Mn,Fe,Co和Ni)及其复合化合物(氧化物,氢氧化物和羟基氧化物)是多种技术的重要材料平台,其中涉及电极电位和溶液pH的电化学相图可用于有效了解其在相关水性环境中的腐蚀和氧化行为。许多以前使用数十年的mTM–Pourbaix图与各种直接电化学观察结果不一致,因为实验复杂性与提取可靠的形成自由能(Δf G)会导致用于建模的数据不准确。在这里,我们开发了一种基于密度泛函理论(DFT)的高通量模拟方法,该方法可以使用高效的DFT方法快速筛选结构和化合物,并使用高级交换相关函数来获得从头算起的精确Δf G值。与过去几十年来报道的各种重要的电化学,地质和生物磁观测结果完全一致的Pourbaix图。我们还分析了控制Δf G之间化学趋势的微观机制值和Pourbaix图可进一步了解基于mTM的材料的电化学行为。最后,我们提供了在可变电极电势和溶液pH值下的概率分布图,以定量显示多相区域和扩散相边界的可能共存。


























 京公网安备 11010802027423号
京公网安备 11010802027423号