当前位置:
X-MOL 学术
›
Faraday Discuss.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Temperature dependence of the vibrational spectrum of porphycene: a qualitative failure of classical-nuclei molecular dynamics.
Faraday Discussions ( IF 3.3 ) Pub Date : 2019-12-16 , DOI: 10.1039/c9fd00056a Yair Litman 1 , Jörg Behler , Mariana Rossi
Faraday Discussions ( IF 3.3 ) Pub Date : 2019-12-16 , DOI: 10.1039/c9fd00056a Yair Litman 1 , Jörg Behler , Mariana Rossi
Affiliation
|
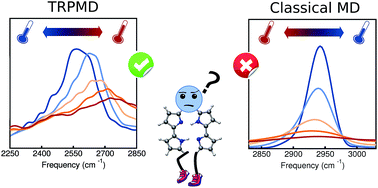
The temperature dependence of vibrational spectra can provide information about structural changes of a system and also serve as a probe to identify different vibrational mode couplings. Fully anharmonic temperature-dependent calculations of these quantities are challenging due to the cost associated with statistically converging trajectory-based methods, especially when accounting for nuclear quantum effects. Here, we train a high-dimensional neural network potential energy surface for the porphycene molecule based on data generated with DFT-B3LYP, including pairwise van der Waals interactions. In addition, we fit a kernel ridge regression model for the molecular dipole moment surface. The combination of this machinery with thermostatted path integral molecular dynamics (TRPMD) allows us to obtain well-converged, full-dimensional, fully-anharmonic vibrational spectra including nuclear quantum effects, without sacrificing the first-principles quality of the potential-energy surface or the dipole surface. Within this framework, we investigate the temperature and isotopologue dependence of the high-frequency vibrational fingerprints of porphycene. While classical-nuclei dynamics predicts a red shift of the vibrations encompassing the NH and CH stretches, TRPMD predicts a strong blue shift in the NH-stretch region and a smaller one in the CH-stretch region. We explain this behavior by analyzing the modulation of the effective potential with temperature, which arises from vibrational coupling between quasi-classical thermally activated modes and high-frequency quantized modes.
中文翻译:

卟啉振动光谱的温度依赖性:经典核分子动力学的定性失效。
振动光谱的温度依赖性可以提供有关系统结构变化的信息,并且还可以用作识别不同振动模式耦合的探针。由于与基于统计轨迹的统计收敛方法相关的成本,尤其是考虑到核量子效应时,这些量的完全非谐温度相关计算是具有挑战性的。在这里,我们基于DFT-B3LYP生成的数据,包括成对的范德华相互作用,为卟啉分子训练了高维神经网络势能面。此外,我们为分子偶极矩表面拟合了核岭回归模型。该机械与恒温路径积分分子动力学(TRPMD)相结合,使我们能够获得收敛良好的全尺寸,包括核量子效应在内的全非谐振动光谱,而不会牺牲势能表面或偶极表面的第一性原理。在此框架内,我们研究了卟啉高频振动指纹图谱对温度和同位素的依赖性。经典核动力学预测包括NH和CH拉伸的振动发生红移,而TRPMD预测NH拉伸区域发生强烈的蓝移,而CH拉伸区域发生较小的蓝移。我们通过分析有效电势随温度的调制来解释这种行为,温度是由准经典热激活模式和高频量化模式之间的振动耦合引起的。而不牺牲势能表面或偶极表面的第一性原理。在此框架内,我们研究了卟啉高频振动指纹图谱对温度和同位素的依赖性。经典核动力学预测包括NH和CH拉伸的振动发生红移,而TRPMD预测NH拉伸区域发生强烈的蓝移,而CH拉伸区域发生较小的蓝移。我们通过分析有效电势随温度的调制来解释这种行为,温度是由准经典热激活模式和高频量化模式之间的振动耦合引起的。而不牺牲势能表面或偶极表面的第一性原理。在此框架内,我们研究了卟啉高频振动指纹图谱对温度和同位素的依赖性。经典核动力学预测包括NH和CH拉伸的振动发生红移,而TRPMD预测NH拉伸区域发生强烈的蓝移,而CH拉伸区域发生较小的蓝移。我们通过分析有效电势随温度的调制来解释这种行为,温度是由准经典热激活模式和高频量化模式之间的振动耦合引起的。我们研究了卟啉高频振动指纹图谱对温度和同位素的依赖性。经典核动力学预测包括NH和CH拉伸的振动发生红移,而TRPMD预测NH拉伸区域发生强烈的蓝移,而CH拉伸区域发生较小的蓝移。我们通过分析有效电势随温度的调制来解释这种行为,温度是由准经典热激活模式和高频量化模式之间的振动耦合引起的。我们研究了卟啉高频振动指纹图谱对温度和同位素的依赖性。经典核动力学预测包括NH和CH拉伸的振动发生红移,而TRPMD预测NH拉伸区域发生强烈的蓝移,而CH拉伸区域发生较小的蓝移。我们通过分析有效电势随温度的调制来解释这种行为,温度是由准经典热激活模式和高频量化模式之间的振动耦合引起的。
更新日期:2019-12-17
中文翻译:
卟啉振动光谱的温度依赖性:经典核分子动力学的定性失效。
振动光谱的温度依赖性可以提供有关系统结构变化的信息,并且还可以用作识别不同振动模式耦合的探针。由于与基于统计轨迹的统计收敛方法相关的成本,尤其是考虑到核量子效应时,这些量的完全非谐温度相关计算是具有挑战性的。在这里,我们基于DFT-B3LYP生成的数据,包括成对的范德华相互作用,为卟啉分子训练了高维神经网络势能面。此外,我们为分子偶极矩表面拟合了核岭回归模型。该机械与恒温路径积分分子动力学(TRPMD)相结合,使我们能够获得收敛良好的全尺寸,包括核量子效应在内的全非谐振动光谱,而不会牺牲势能表面或偶极表面的第一性原理。在此框架内,我们研究了卟啉高频振动指纹图谱对温度和同位素的依赖性。经典核动力学预测包括NH和CH拉伸的振动发生红移,而TRPMD预测NH拉伸区域发生强烈的蓝移,而CH拉伸区域发生较小的蓝移。我们通过分析有效电势随温度的调制来解释这种行为,温度是由准经典热激活模式和高频量化模式之间的振动耦合引起的。而不牺牲势能表面或偶极表面的第一性原理。在此框架内,我们研究了卟啉高频振动指纹图谱对温度和同位素的依赖性。经典核动力学预测包括NH和CH拉伸的振动发生红移,而TRPMD预测NH拉伸区域发生强烈的蓝移,而CH拉伸区域发生较小的蓝移。我们通过分析有效电势随温度的调制来解释这种行为,温度是由准经典热激活模式和高频量化模式之间的振动耦合引起的。而不牺牲势能表面或偶极表面的第一性原理。在此框架内,我们研究了卟啉高频振动指纹图谱对温度和同位素的依赖性。经典核动力学预测包括NH和CH拉伸的振动发生红移,而TRPMD预测NH拉伸区域发生强烈的蓝移,而CH拉伸区域发生较小的蓝移。我们通过分析有效电势随温度的调制来解释这种行为,温度是由准经典热激活模式和高频量化模式之间的振动耦合引起的。我们研究了卟啉高频振动指纹图谱对温度和同位素的依赖性。经典核动力学预测包括NH和CH拉伸的振动发生红移,而TRPMD预测NH拉伸区域发生强烈的蓝移,而CH拉伸区域发生较小的蓝移。我们通过分析有效电势随温度的调制来解释这种行为,温度是由准经典热激活模式和高频量化模式之间的振动耦合引起的。我们研究了卟啉高频振动指纹图谱对温度和同位素的依赖性。经典核动力学预测包括NH和CH拉伸的振动发生红移,而TRPMD预测NH拉伸区域发生强烈的蓝移,而CH拉伸区域发生较小的蓝移。我们通过分析有效电势随温度的调制来解释这种行为,温度是由准经典热激活模式和高频量化模式之间的振动耦合引起的。










































 京公网安备 11010802027423号
京公网安备 11010802027423号