当前位置:
X-MOL 学术
›
Mol. Omics
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Prediction of zinc-binding sites using multiple sequence profiles and machine learning methods.
Molecular Omics ( IF 3.0 ) Pub Date : 2019-05-02 , DOI: 10.1039/c9mo00043g Renxiang Yan 1 , Xiaofeng Wang 2 , Yarong Tian 3 , Jing Xu 1 , Xiaoli Xu 4 , Juan Lin 1
Molecular Omics ( IF 3.0 ) Pub Date : 2019-05-02 , DOI: 10.1039/c9mo00043g Renxiang Yan 1 , Xiaofeng Wang 2 , Yarong Tian 3 , Jing Xu 1 , Xiaoli Xu 4 , Juan Lin 1
Affiliation
|
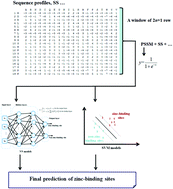
The zinc (Zn2+) cofactor has been proven to be involved in numerous biological mechanisms and the zinc-binding site is recognized as one of the most important post-translation modifications in proteins. Therefore, accurate knowledge of zinc ions in protein structures can provide potential clues for elucidation of protein folding and functions. However, determining zinc-binding residues by experimental means is usually lab-intensive and associated with high cost in most cases. In this context, the development of computational tools for identifying zinc-binding sites is highly desired, especially in the current post-genomic era. In this work, we developed a novel zinc-binding site prediction method by combining several intensively-trained machine learning models. To establish an accurate and generative method, we downloaded all zinc-binding proteins from the Protein Data Bank and prepared a non-redundant dataset. Meanwhile, a well-prepared dataset by other groups was also used. Then, effective and complementary features were extracted from sequences and three-dimensional structures of these proteins. Moreover, several well-designed machine learning models were intensively trained to construct accurate models. To assess the performance, the obtained predictors were stringently benchmarked using the diverse zinc-binding sites. Furthermore, several state-of-the-art in silico methods developed specifically for zinc-binding sites were also evaluated and compared. The results confirmed that our method is very competitive in real world applications and could become a complementary tool to wet lab experiments. To facilitate research in the community, a web server and stand-alone program implementing our method were constructed and are publicly available at . The downloadable program of our method can be easily used for the high-throughput screening of potential zinc-binding sites across proteomes.
中文翻译:

使用多个序列图谱和机器学习方法预测锌结合位点。
锌(Zn2 +)辅助因子已被证明参与多种生物学机制,并且锌结合位点被认为是蛋白质中最重要的翻译后修饰之一。因此,准确了解蛋白质结构中的锌离子可为阐明蛋白质折叠和功能提供潜在的线索。然而,通过实验手段确定锌结合残基通常是实验室密集的,并且在大多数情况下与高成本有关。在这种情况下,非常需要开发用于识别锌结合位点的计算工具,尤其是在当前的后基因组时代。在这项工作中,我们通过结合几种经过严格训练的机器学习模型,开发了一种新颖的锌结合位点预测方法。要建立一种准确而有创见的方法,我们从蛋白质数据库下载了所有锌结合蛋白,并准备了一个非冗余数据集。同时,还使用了其他小组精心准备的数据集。然后,从这些蛋白质的序列和三维结构中提取有效和互补的特征。此外,对数种精心设计的机器学习模型进行了严格的训练,以构建准确的模型。为了评估性能,使用各种锌结合位点对获得的预测因子进行了严格的基准测试。此外,还评估并比较了几种专门为锌结合位点开发的计算机技术。结果证实,我们的方法在实际应用中具有很大的竞争力,并且可以成为湿实验室实验的补充工具。为了促进社区研究,已构建了实现我们方法的Web服务器和独立程序,并在公开提供了该程序。我们方法的可下载程序可轻松用于蛋白质组中潜在锌结合位点的高通量筛选。
更新日期:2019-06-11
中文翻译:
使用多个序列图谱和机器学习方法预测锌结合位点。
锌(Zn2 +)辅助因子已被证明参与多种生物学机制,并且锌结合位点被认为是蛋白质中最重要的翻译后修饰之一。因此,准确了解蛋白质结构中的锌离子可为阐明蛋白质折叠和功能提供潜在的线索。然而,通过实验手段确定锌结合残基通常是实验室密集的,并且在大多数情况下与高成本有关。在这种情况下,非常需要开发用于识别锌结合位点的计算工具,尤其是在当前的后基因组时代。在这项工作中,我们通过结合几种经过严格训练的机器学习模型,开发了一种新颖的锌结合位点预测方法。要建立一种准确而有创见的方法,我们从蛋白质数据库下载了所有锌结合蛋白,并准备了一个非冗余数据集。同时,还使用了其他小组精心准备的数据集。然后,从这些蛋白质的序列和三维结构中提取有效和互补的特征。此外,对数种精心设计的机器学习模型进行了严格的训练,以构建准确的模型。为了评估性能,使用各种锌结合位点对获得的预测因子进行了严格的基准测试。此外,还评估并比较了几种专门为锌结合位点开发的计算机技术。结果证实,我们的方法在实际应用中具有很大的竞争力,并且可以成为湿实验室实验的补充工具。为了促进社区研究,已构建了实现我们方法的Web服务器和独立程序,并在公开提供了该程序。我们方法的可下载程序可轻松用于蛋白质组中潜在锌结合位点的高通量筛选。










































 京公网安备 11010802027423号
京公网安备 11010802027423号