当前位置:
X-MOL 学术
›
ACS Pharmacol. Transl. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Differential Sensitivity to CDK2 Inhibition Discriminates the Molecular Mechanisms of CHK1 Inhibitors as Monotherapy or in Combination with the Topoisomerase I Inhibitor SN38.
ACS Pharmacology & Translational Science ( IF 4.9 ) Pub Date : 2019-04-12 , DOI: 10.1021/acsptsci.9b00001 Nicholas J H Warren 1 , Katelyn L Donahue 1 , Alan Eastman 1
ACS Pharmacology & Translational Science ( IF 4.9 ) Pub Date : 2019-04-12 , DOI: 10.1021/acsptsci.9b00001 Nicholas J H Warren 1 , Katelyn L Donahue 1 , Alan Eastman 1
Affiliation
|
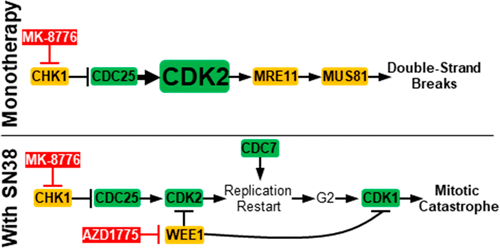
DNA damage activates checkpoints to arrest cell cycle progression in S and G2 phases, thereby providing time for repair and recovery. The combination of DNA-damaging agents and inhibitors of CHK1 (CHK1i) is an emerging strategy for sensitizing cancer cells. CHK1i induce replication on damaged DNA and mitosis before repair is complete, and this occurs in a majority of cell lines. However, ∼15% of cancer cell lines are hypersensitive to single-agent CHK1i. As both abrogation of S phase arrest and single-agent activity depend on CDK2, this study resolved how activation of CDK2 can be essential for both replication and cytotoxicity. S phase arrest was induced with the topoisomerase I inhibitor SN38; the addition of CHK1i rapidly activated CDK2, inducing S phase progression that was inhibited by the CDK2 inhibitor CVT-313. In contrast, DNA damage and cytotoxicity induced by single-agent CHK1i in hypersensitive cell lines were also inhibited by CVT-313 but at 20-fold lower concentrations. The differential sensitivity to CVT-313 is explained by different activity thresholds required for phosphorylation of CDK2 substrates. While the critical CDK2 substrates are not yet defined, we conclude that hypersensitivity to single-agent CHK1i depends on phosphorylation of substrates that require high CDK2 activity levels. Surprisingly, CHK1i did not increase SN38-mediated cytotoxicity. In contrast, while inhibition of WEE1 also abrogated S phase arrest, it more directly activated CDK1, induced premature mitosis, and enhanced cytotoxicity. Hence, while high activity of CDK2 is critical for cytotoxicity of single-agent CHK1i, CDK1 is additionally required for sensitivity to the drug combination.
中文翻译:

对CDK2抑制的敏感性不同,可以区分CHK1抑制剂作为单一疗法或与拓扑异构酶I抑制剂SN38组合的分子机制。
DNA损伤会激活检查点,以阻止S和G2期的细胞周期进程,从而为修复和恢复提供时间。DNA破坏剂和CHK1抑制剂(CHK1i)的组合是致敏癌细胞的新兴策略。CHK1i会在修复完成之前诱导受损DNA的复制和有丝分裂,这在大多数细胞系中都会发生。但是,约有15%的癌细胞系对单药CHK1i过敏。由于S期停滞和单药活性的废除都取决于CDK2,因此本研究解决了CDK2活化对于复制和细胞毒性必不可少的问题。拓扑异构酶I抑制剂SN38诱导S期阻滞;CHK1i的添加会迅速激活CDK2,诱导被CDK2抑制剂CVT-313抑制的S期进程。相比之下,CVT-313也可抑制超敏细胞株中单药CHK1i诱导的DNA损伤和细胞毒性,但浓度要低20倍。CDV2底物磷酸化所需的不同活性阈值解释了对CVT-313的敏感性差异。尽管尚未定义关键的CDK2底物,但我们得出的结论是,对单药CHK1i的超敏反应取决于需要高CDK2活性水平的底物的磷酸化。令人惊讶的是,CHK1i没有增加SN38介导的细胞毒性。相反,虽然对WEE1的抑制也废除了S期阻滞,但它更直接地激活CDK1,诱导过早有丝分裂,并增强了细胞毒性。因此,尽管CDK2的高活性对于单药CHK1i的细胞毒性至关重要,
更新日期:2019-05-23
中文翻译:
对CDK2抑制的敏感性不同,可以区分CHK1抑制剂作为单一疗法或与拓扑异构酶I抑制剂SN38组合的分子机制。
DNA损伤会激活检查点,以阻止S和G2期的细胞周期进程,从而为修复和恢复提供时间。DNA破坏剂和CHK1抑制剂(CHK1i)的组合是致敏癌细胞的新兴策略。CHK1i会在修复完成之前诱导受损DNA的复制和有丝分裂,这在大多数细胞系中都会发生。但是,约有15%的癌细胞系对单药CHK1i过敏。由于S期停滞和单药活性的废除都取决于CDK2,因此本研究解决了CDK2活化对于复制和细胞毒性必不可少的问题。拓扑异构酶I抑制剂SN38诱导S期阻滞;CHK1i的添加会迅速激活CDK2,诱导被CDK2抑制剂CVT-313抑制的S期进程。相比之下,CVT-313也可抑制超敏细胞株中单药CHK1i诱导的DNA损伤和细胞毒性,但浓度要低20倍。CDV2底物磷酸化所需的不同活性阈值解释了对CVT-313的敏感性差异。尽管尚未定义关键的CDK2底物,但我们得出的结论是,对单药CHK1i的超敏反应取决于需要高CDK2活性水平的底物的磷酸化。令人惊讶的是,CHK1i没有增加SN38介导的细胞毒性。相反,虽然对WEE1的抑制也废除了S期阻滞,但它更直接地激活CDK1,诱导过早有丝分裂,并增强了细胞毒性。因此,尽管CDK2的高活性对于单药CHK1i的细胞毒性至关重要,










































 京公网安备 11010802027423号
京公网安备 11010802027423号