npj Genomic Medicine ( IF 4.7 ) Pub Date : 2019-02-01 , DOI: 10.1038/s41525-019-0077-8 Shicai Fan , Jianxiong Tang , Nan Li , Ying Zhao , Rizi Ai , Kai Zhang , Mengchi Wang , Wei Du , Wei Wang
|
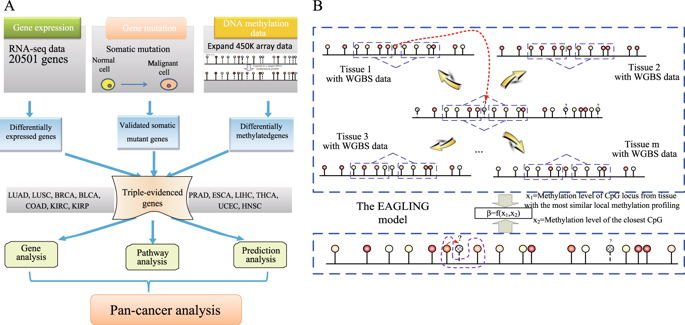
The integration of genomic and DNA methylation data has been demonstrated as a powerful strategy in understanding cancer mechanisms and identifying therapeutic targets. The TCGA consortium has mapped DNA methylation in thousands of cancer samples using Illumina Infinium Human Methylation 450 K BeadChip (Illumina 450 K array) that only covers about 1.5% of CpGs in the human genome. Therefore, increasing the coverage of the DNA methylome would significantly leverage the usage of the TCGA data. Here, we present a new model called EAGLING that can expand the Illumina 450 K array data 18 times to cover about 30% of the CpGs in the human genome. We applied it to analyze 13 cancers in TCGA. By integrating the expanded methylation, gene expression, and somatic mutation data, we identified the genes showing differential patterns in each of the 13 cancers. Many of the triple-evidenced genes identified in majority of the cancers are biomarkers or potential biomarkers. Pan-cancer analysis also revealed the pathways in which the triple-evidenced genes are enriched, which include well known ones as well as new ones, such as axonal guidance signaling pathway and pathways related to inflammatory processing or inflammation response. Triple-evidenced genes, particularly TNXB, RRM2, CELSR3, SLC16A3, FANCI, MMP9, MMP11, SIK1, and TRIM59 showed superior predictive power in both tumor diagnosis and prognosis. These results have demonstrated that the integrative analysis using the expanded methylation data is powerful in identifying critical genes/pathways that may serve as new therapeutic targets.
中文翻译:
结合扩展的DNA甲基化数据进行的综合分析揭示了癌症中常见的关键调节剂和途径
基因组和DNA甲基化数据的整合已被证明是了解癌症机制和确定治疗靶点的有力策略。TCGA联盟已使用Illumina Infinium Human Methylation 450 K BeadChip(Illumina 450 K阵列)绘制了数千个癌症样品中的DNA甲基化图,该芯片仅覆盖人类基因组中约1.5%的CpG。因此,增加DNA甲基化组的覆盖范围将大大利用TCGA数据的使用。在这里,我们提出了一种名为EAGLING的新模型,该模型可以将Illumina 450 K阵列数据扩展18倍,以覆盖人类基因组中约30%的CpG。我们将其用于分析TCGA中的13种癌症。通过整合扩展的甲基化,基因表达和体细胞突变数据,我们确定了在13种癌症中每种均表现出差异模式的基因。在大多数癌症中鉴定出的许多三证基因是生物标志物或潜在的生物标志物。泛癌分析还揭示了三证基因富集的途径,其中既包括众所周知的基因,也包括新的基因,例如轴突指导信号转导途径以及与炎症过程或炎症反应相关的途径。三重证据基因,特别是TNXB,RRM2,CELSR3,SLC16A3,FANCI,MMP9,MMP11,SIK1和TRIM59在肿瘤诊断和预后方面均显示出优异的预测能力。这些结果表明,使用扩展的甲基化数据进行的综合分析在确定可用作新治疗靶标的关键基因/途径方面功能强大。泛癌分析还揭示了三证基因富集的途径,其中既包括众所周知的基因,也包括新的基因,例如轴突指导信号转导途径以及与炎症过程或炎症反应相关的途径。三重证据基因,特别是TNXB,RRM2,CELSR3,SLC16A3,FANCI,MMP9,MMP11,SIK1和TRIM59在肿瘤诊断和预后方面均显示出优异的预测能力。这些结果表明,使用扩展的甲基化数据进行的综合分析在确定可用作新治疗靶标的关键基因/途径方面功能强大。泛癌分析还揭示了三证基因富集的途径,其中既包括众所周知的基因,也包括新的基因,例如轴突指导信号转导途径以及与炎症过程或炎症反应相关的途径。三重证据基因,特别是TNXB,RRM2,CELSR3,SLC16A3,FANCI,MMP9,MMP11,SIK1和TRIM59在肿瘤诊断和预后方面均显示出优异的预测能力。这些结果表明,使用扩展的甲基化数据进行的综合分析在确定可用作新治疗靶标的关键基因/途径方面功能强大。例如轴突引导信号转导途径以及与炎症过程或炎症反应相关的途径。三重证据基因,特别是TNXB,RRM2,CELSR3,SLC16A3,FANCI,MMP9,MMP11,SIK1和TRIM59在肿瘤诊断和预后方面均显示出优异的预测能力。这些结果表明,使用扩展的甲基化数据进行的综合分析在确定可用作新治疗靶标的关键基因/途径方面功能强大。例如轴突引导信号转导途径以及与炎症过程或炎症反应相关的途径。三重证据基因,特别是TNXB,RRM2,CELSR3,SLC16A3,FANCI,MMP9,MMP11,SIK1和TRIM59在肿瘤诊断和预后方面均显示出优异的预测能力。这些结果表明,使用扩展的甲基化数据进行的综合分析在确定可用作新治疗靶标的关键基因/途径方面功能强大。










































 京公网安备 11010802027423号
京公网安备 11010802027423号