当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Is AMOEBA a Good Force Field for Molecular Dynamics Simulations of Carbohydrates?
Journal of Chemical Information and Modeling ( IF 5.3 ) Pub Date : 2025-05-20 , DOI: 10.1021/acs.jcim.5c00442 Mawuli Deegbey , Ethan W. Sumner , Valerie Vaissier Welborn
Journal of Chemical Information and Modeling ( IF 5.3 ) Pub Date : 2025-05-20 , DOI: 10.1021/acs.jcim.5c00442 Mawuli Deegbey , Ethan W. Sumner , Valerie Vaissier Welborn
|
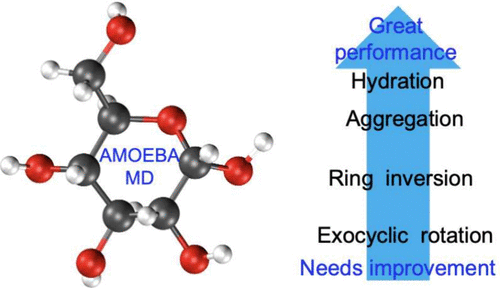
Over the years, molecular dynamics (MD) simulations have been employed in the study of carbohydrates, with force fields such as CHARMM, AMBER/GLYCAM, and GROMOS. Although these force fields have achieved considerable success and played a pivotal role in our understanding of carbohydrate chemistry, growing interest has emerged in incorporating polarization effects to enhance the accuracy of simulations. In this perspective, we contemplate the advances that have been made in nonpolarizable and polarizable force fields to extract the key factors controlling accuracy in MD of carbohydrates. We find that the extreme hydrophilicity and conformational flexibility of carbohydrates pose challenges for most force fields. Overall, a force field suited for carbohydrates needs to include a water model developed consistently with the solute parameter sets, a soft van der Waals repulsion term at short distances, and polarization (whether implicit or explicit). We find that AMOEBA improves the prediction of hydration shell structure and dynamics, hydrogen bonding, and kinetics of diffusion, although it remains largely untested for conformational flexibility and glycosidic linkages. Nevertheless, AMOEBA's recent success in modeling monosaccharides without revisions of the potential energy functions or water model presents a promising avenue for future research. Such advances will provide deeper insights into the structure, dynamics, and interactions of these biologically and industrially relevant macromolecules.
中文翻译:

AMOEBA 是碳水化合物分子动力学模拟的良好力场吗?
多年来,分子动力学 (MD) 模拟一直被用于碳水化合物的研究,包括 CHARMM、AMBER/GLYCAM 和 GRROMOS 等力场。尽管这些力场已经取得了相当大的成功,并在我们对碳水化合物化学的理解中发挥了关键作用,但人们对结合极化效应以提高模拟的准确性的兴趣越来越大。从这个角度来看,我们考虑了在非极化和极化力场方面取得的进展,以提取控制碳水化合物 MD 准确性的关键因素。我们发现碳水化合物的极端亲水性和构象灵活性对大多数力场构成了挑战。总的来说,适合碳水化合物的力场需要包括一个与溶质参数集一致开发的水模型、短距离的软范德华排斥项和极化(无论是隐式还是显式)。我们发现 AMOEBA 改善了对水合壳结构和动力学、氢键和扩散动力学的预测,尽管它在很大程度上仍未测试构象灵活性和糖苷键。尽管如此,AMOEBA 最近在没有修改势能函数或水模型的情况下成功地对单糖进行建模,这为未来的研究提供了一条有希望的途径。这些进展将为这些生物和工业相关大分子的结构、动力学和相互作用提供更深入的见解。
更新日期:2025-05-20
中文翻译:
AMOEBA 是碳水化合物分子动力学模拟的良好力场吗?
多年来,分子动力学 (MD) 模拟一直被用于碳水化合物的研究,包括 CHARMM、AMBER/GLYCAM 和 GRROMOS 等力场。尽管这些力场已经取得了相当大的成功,并在我们对碳水化合物化学的理解中发挥了关键作用,但人们对结合极化效应以提高模拟的准确性的兴趣越来越大。从这个角度来看,我们考虑了在非极化和极化力场方面取得的进展,以提取控制碳水化合物 MD 准确性的关键因素。我们发现碳水化合物的极端亲水性和构象灵活性对大多数力场构成了挑战。总的来说,适合碳水化合物的力场需要包括一个与溶质参数集一致开发的水模型、短距离的软范德华排斥项和极化(无论是隐式还是显式)。我们发现 AMOEBA 改善了对水合壳结构和动力学、氢键和扩散动力学的预测,尽管它在很大程度上仍未测试构象灵活性和糖苷键。尽管如此,AMOEBA 最近在没有修改势能函数或水模型的情况下成功地对单糖进行建模,这为未来的研究提供了一条有希望的途径。这些进展将为这些生物和工业相关大分子的结构、动力学和相互作用提供更深入的见解。






















































 京公网安备 11010802027423号
京公网安备 11010802027423号