当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Benchmark Investigation of SCC-DFTB against Standard and Hybrid DFT to Model Electronic Properties in Two-Dimensional MOFs for Thermoelectric Applications
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2024-05-06 , DOI: 10.1021/acs.jctc.3c01405 Masoumeh Mahmoudi Gahrouei 1 , Nikiphoros Vlastos 1 , Ransell D’Souza 2 , Emmanuel C. Odogwu 1 , Laura de Sousa Oliveira 1
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2024-05-06 , DOI: 10.1021/acs.jctc.3c01405 Masoumeh Mahmoudi Gahrouei 1 , Nikiphoros Vlastos 1 , Ransell D’Souza 2 , Emmanuel C. Odogwu 1 , Laura de Sousa Oliveira 1
Affiliation
|
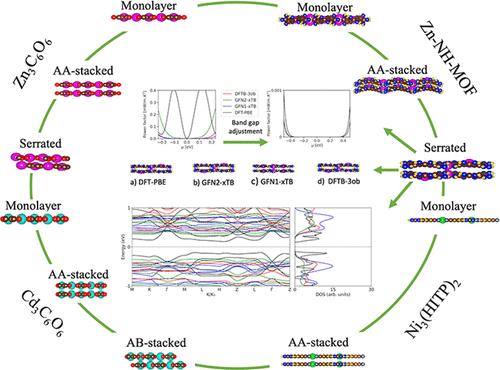
Recent studies have shown that metal−organic frameworks (MOFs) have potential as thermoelectric materials, and the topic has received increasing attention. The main motivation for this project is to further our knowledge of thermoelectric properties in MOFs and find which available self-consistent-charge density functional tight binding (SCC-DFTB) method can best predict (at least trends in) the electronic properties of MOFs at a lower computational cost than standard density functional theory (DFT). In this work, the electronic properties of monolayer, serrated, AA-stacked, and/or AB-stacked Zn3C6O6, Cd3C6O6, Zn-NH-MOF─for which no previous calculations of thermoelectric performance exist─and Ni3(HITP)2 MOFs are modeled with DFT-PBE, DFT-HSE06, GFN1-xTB, GFN2-xTB, and DFTB-3ob/mio. The band structures, density of states, and their relative orbital contributions, as well as the electrical conductivity, Seebeck coefficient, and power factor, are compared across methods and geometries. Our results suggest that GFN-xTB is adequate to predict the MOFs’ band structure shape and density of states but not band gap. Our calculations further indicate that Zn3C6O6, Cd3C6O6, and Zn-NH-MOF have higher power factor values than Ni3(HITP)2, one of the highest performing synthesized MOFs, and are therefore promising for thermoelectric applications.
中文翻译:

SCC-DFTB 与标准和混合 DFT 的基准研究,用于模拟热电应用二维 MOF 的电子特性
最近的研究表明,金属有机框架(MOF)具有作为热电材料的潜力,并且该主题受到越来越多的关注。该项目的主要动机是加深我们对 MOF 热电特性的了解,并找到哪种可用的自洽电荷密度功能紧密结合 (SCC-DFTB) 方法可以最好地预测 MOF 的电子特性(至少是趋势)比标准密度泛函理论 (DFT) 的计算成本更低。在这项工作中,单层、锯齿状、AA 堆叠和/或 AB 堆叠 Zn 3 C 6 O 6、Cd 3 C 6 O 6、Zn-NH-MOF 的电子特性 — 之前没有计算过热电性能存在 — 并且 Ni 3 (HITP) 2 MOF 使用 DFT-PBE、DFT-HSE06、GFN1-xTB、GFN2-xTB 和 DFTB-3ob/mio 进行建模。不同方法和几何结构对能带结构、态密度及其相对轨道贡献以及电导率、塞贝克系数和功率因数进行了比较。我们的结果表明 GFN-xTB 足以预测 MOF 的能带结构形状和态密度,但不能预测带隙。我们的计算进一步表明,Zn 3 C 6 O 6、Cd 3 C 6 O 6和 Zn-NH-MOF 具有比性能最高的合成 MOF 之一 Ni 3 (HITP) 2更高的功率因数值,因此很有前景用于热电应用。
更新日期:2024-05-06
中文翻译:
SCC-DFTB 与标准和混合 DFT 的基准研究,用于模拟热电应用二维 MOF 的电子特性
最近的研究表明,金属有机框架(MOF)具有作为热电材料的潜力,并且该主题受到越来越多的关注。该项目的主要动机是加深我们对 MOF 热电特性的了解,并找到哪种可用的自洽电荷密度功能紧密结合 (SCC-DFTB) 方法可以最好地预测 MOF 的电子特性(至少是趋势)比标准密度泛函理论 (DFT) 的计算成本更低。在这项工作中,单层、锯齿状、AA 堆叠和/或 AB 堆叠 Zn 3 C 6 O 6、Cd 3 C 6 O 6、Zn-NH-MOF 的电子特性 — 之前没有计算过热电性能存在 — 并且 Ni 3 (HITP) 2 MOF 使用 DFT-PBE、DFT-HSE06、GFN1-xTB、GFN2-xTB 和 DFTB-3ob/mio 进行建模。不同方法和几何结构对能带结构、态密度及其相对轨道贡献以及电导率、塞贝克系数和功率因数进行了比较。我们的结果表明 GFN-xTB 足以预测 MOF 的能带结构形状和态密度,但不能预测带隙。我们的计算进一步表明,Zn 3 C 6 O 6、Cd 3 C 6 O 6和 Zn-NH-MOF 具有比性能最高的合成 MOF 之一 Ni 3 (HITP) 2更高的功率因数值,因此很有前景用于热电应用。































 京公网安备 11010802027423号
京公网安备 11010802027423号