当前位置:
X-MOL 学术
›
J. Phys. Chem. B
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Toward Modeling the Structure of Electrolytes at Charged Mineral Interfaces Using Classical Density Functional Theory
The Journal of Physical Chemistry B ( IF 3.3 ) Pub Date : 2024-04-16 , DOI: 10.1021/acs.jpcb.3c08045 Thomas Petersen 1
The Journal of Physical Chemistry B ( IF 3.3 ) Pub Date : 2024-04-16 , DOI: 10.1021/acs.jpcb.3c08045 Thomas Petersen 1
Affiliation
|
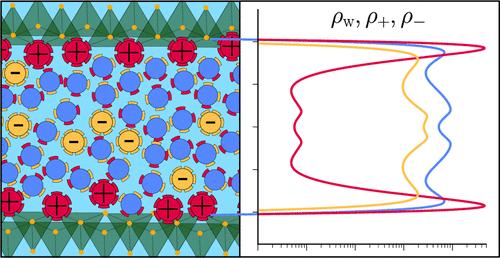
The organization of water molecules and ions between charged mineral surfaces determines the stability of colloidal suspensions and the strength of phase-separated particulate gels. In this article, we assemble a density functional that measures the free energy due to the interaction of water molecules and ions in electric double layers. The model accounts for the finite size of the particles using fundamental measure theory, hydrogen-bonding between water molecules using Wertheim’s statistical association theory, long-range dispersion interactions using Barker and Henderson’s high-temperature expansion, electrostatic correlations using a functionalized mean-spherical approximation, and Coulomb forces through the Poisson equation. These contributions are shown to produce highly correlated structures, aptly rendering the layering of counterions and co-ions at highly charged surfaces and permitting the solvation of ions and surfaces to be measured by a combination of short-range associations and long-ranged attractions. The model is tested in a planar geometry near soft, charged surfaces to reproduce the structure of water near graphene and mica. For mica surfaces, explicitly representing the density of the outer oxygen layer of the exposed silica tetrahedra allows water molecules to hydrogen-bond to the solid. When electrostatic interactions are included, water molecules assume a hybrid character, being accounted for implicitly in the dielectric constant but explicitly otherwise. The disjoining pressure between approaching like-charged surfaces is calculated, demonstrating the model’s ability to probe pressure oscillations that arise during the expulsion of ions and water layers from the interfacial gap and predict strong interattractive stresses that form at narrow interfacial spacing when the surface charge is overscreened. This interattractive stress arises not due to in-plane correlations under strong electrostatic coupling but due to the out-of-plane structuring of associating ions and water molecules.
中文翻译:

使用经典密度泛函理论对带电矿物界面处的电解质结构进行建模
带电矿物表面之间的水分子和离子的组织决定了胶体悬浮液的稳定性和相分离颗粒凝胶的强度。在本文中,我们组装了一个密度泛函,用于测量由于双电层中水分子和离子相互作用而产生的自由能。该模型使用基本测量理论解释颗粒的有限尺寸,使用 Wertheim 统计关联理论解释水分子之间的氢键,使用 Barker 和 Henderson 的高温膨胀解释长程色散相互作用,使用功能化平均球近似解释静电关联,以及通过泊松方程得到的库仑力。这些贡献被证明可以产生高度相关的结构,适当地在高电荷表面呈现反离子和共离子的分层,并允许通过短程缔合和长程吸引力的组合来测量离子和表面的溶剂化。该模型在柔软、带电表面附近的平面几何结构中进行了测试,以重现石墨烯和云母附近的水结构。对于云母表面,明确表示暴露的二氧化硅四面体的外部氧层的密度允许水分子与固体形成氢键。当包括静电相互作用时,水分子呈现混合特征,在介电常数中隐含地解释了这一点,但在其他方面则明确地解释了这一点。计算接近的带电表面之间的分离压力,证明该模型能够探测离子和水层从界面间隙排出过程中产生的压力振荡,并预测当表面电荷处于狭窄界面间距时形成的强相互作用应力。过度筛选。这种相互吸引力的产生不是由于强静电耦合下的面内相关性,而是由于缔合离子和水分子的面外结构。
更新日期:2024-04-16
中文翻译:
使用经典密度泛函理论对带电矿物界面处的电解质结构进行建模
带电矿物表面之间的水分子和离子的组织决定了胶体悬浮液的稳定性和相分离颗粒凝胶的强度。在本文中,我们组装了一个密度泛函,用于测量由于双电层中水分子和离子相互作用而产生的自由能。该模型使用基本测量理论解释颗粒的有限尺寸,使用 Wertheim 统计关联理论解释水分子之间的氢键,使用 Barker 和 Henderson 的高温膨胀解释长程色散相互作用,使用功能化平均球近似解释静电关联,以及通过泊松方程得到的库仑力。这些贡献被证明可以产生高度相关的结构,适当地在高电荷表面呈现反离子和共离子的分层,并允许通过短程缔合和长程吸引力的组合来测量离子和表面的溶剂化。该模型在柔软、带电表面附近的平面几何结构中进行了测试,以重现石墨烯和云母附近的水结构。对于云母表面,明确表示暴露的二氧化硅四面体的外部氧层的密度允许水分子与固体形成氢键。当包括静电相互作用时,水分子呈现混合特征,在介电常数中隐含地解释了这一点,但在其他方面则明确地解释了这一点。计算接近的带电表面之间的分离压力,证明该模型能够探测离子和水层从界面间隙排出过程中产生的压力振荡,并预测当表面电荷处于狭窄界面间距时形成的强相互作用应力。过度筛选。这种相互吸引力的产生不是由于强静电耦合下的面内相关性,而是由于缔合离子和水分子的面外结构。


























 京公网安备 11010802027423号
京公网安备 11010802027423号