当前位置:
X-MOL 学术
›
Comp. Mater. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Tuning structure, stability and magnetism in vanadium doped zinc sulfide for spintronic applications: Insights from first-principles and Monte Carlo approaches
Computational Materials Science ( IF 3.3 ) Pub Date : 2024-04-06 , DOI: 10.1016/j.commatsci.2024.112995 Abdelhamid Ait M’hid , Guojian Li , Mourad Boughrara , Mohamed Kerouad , Qiang Wang
Computational Materials Science ( IF 3.3 ) Pub Date : 2024-04-06 , DOI: 10.1016/j.commatsci.2024.112995 Abdelhamid Ait M’hid , Guojian Li , Mourad Boughrara , Mohamed Kerouad , Qiang Wang
|
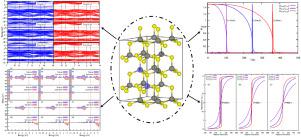
This work utilizes a multi-scale computational approach combining first-principles density functional theory (DFT) and Monte Carlo statistical simulations to provide fundamental insights into the impacts of vanadium (V) doping on the structural, electronic, magnetic, and thermodynamic properties of wurtzite zinc sulfide (ZnS). 2 × 2 × 2 ZnS supercells with controlled V doping concentrations from 12.5%–25% substituting zinc sites were constructed. The introduction of V dopants induces systematic expansions in lattice parameters and cell volume, attributed to the larger ionic radius of V than Zn, while the average compressibility shows minimal variation. However, V incorporation is found to soften ZnS by over 15% at higher dopant contents, increasing compliance and anisotropy. Stable ferromagnetic spin couplings emerge, mediated by host carriers without precipitating V secondary phases. The band structure develops half-metallic electronic properties with the lifting of spin degeneracy and preferential spin-up band metallicity induced by V-3d and S-3p hybridization effects. This results in 100% spin polarization at the Fermi level. Monte Carlo simulations based on a tight-binding Hamiltonian constructed from maximally localized Wannier functions to accurately calculate the exchange interactions between V spins successfully reproduce the magnetic phase transition behavior. The results confirm high Curie temperatures exceeding 300 K at 25% V, attributable to the significant strengthening of short-range ferromagnetic couplings over antiferromagnetic interactions. The integrated computational approach provides fundamental insights into the microscopic origin of carrier-mediated ferromagnetism in V-doped wurtzite ZnS dilute magnetic semiconductors. The systematic first-principles calculations and statistical mechanics modeling establish clear structure–property relationships linking V doping configurations and concentrations to tuned mechanical response, enhanced stability, electronic structure modifications, spin polarization, and magnetic ordering temperatures in ZnS. The results present design pathways and predictive capabilities to guide experimental efforts toward emerging high-performance magnetic materials for spintronic technologies.
中文翻译:

用于自旋电子学应用的钒掺杂硫化锌的结构、稳定性和磁性的调节:第一性原理和蒙特卡罗方法的见解
这项工作利用多尺度计算方法,结合第一原理密度泛函理论 (DFT) 和蒙特卡罗统计模拟,为钒 (V) 掺杂对纤锌矿结构、电子、磁性和热力学性质的影响提供基本见解硫化锌(ZnS)。构建了 2 × 2 × 2 ZnS 超级电池,其 V 掺杂浓度受控为 12.5%–25% 替代锌位点。 V掺杂剂的引入导致晶格参数和晶胞体积的系统膨胀,这归因于V的离子半径比Zn更大,而平均压缩率显示出最小的变化。然而,人们发现,在较高掺杂剂含量下,V 的掺入可使 ZnS 软化超过 15%,从而增加柔量和各向异性。稳定的铁磁自旋耦合出现,由主载流子介导,而不沉淀 V 第二相。该能带结构随着 V-3d 和 S-3p 杂化效应引起的自旋简并性和优先自旋能带金属性的提升而发展出半金属电子特性。这导致费米能级处 100% 自旋极化。基于由最大局域 Wannier 函数构建的紧束缚哈密顿量的蒙特卡罗模拟能够准确计算 V 自旋之间的交换相互作用,成功地再现了磁相变行为。结果证实,25% V 时居里温度超过 300 K,这归因于短程铁磁耦合相对于反铁磁相互作用的显着增强。集成计算方法为掺杂 V 纤锌矿 ZnS 稀磁半导体中载流子介导的铁磁性的微观起源提供了基本见解。系统的第一性原理计算和统计力学模型建立了清晰的结构-性能关系,将 V 掺杂构型和浓度与 ZnS 中的机械响应、稳定性增强、电子结构修饰、自旋极化和磁有序温度联系起来。研究结果提出了设计途径和预测能力,以指导自旋电子技术的新兴高性能磁性材料的实验工作。
更新日期:2024-04-06
中文翻译:
用于自旋电子学应用的钒掺杂硫化锌的结构、稳定性和磁性的调节:第一性原理和蒙特卡罗方法的见解
这项工作利用多尺度计算方法,结合第一原理密度泛函理论 (DFT) 和蒙特卡罗统计模拟,为钒 (V) 掺杂对纤锌矿结构、电子、磁性和热力学性质的影响提供基本见解硫化锌(ZnS)。构建了 2 × 2 × 2 ZnS 超级电池,其 V 掺杂浓度受控为 12.5%–25% 替代锌位点。 V掺杂剂的引入导致晶格参数和晶胞体积的系统膨胀,这归因于V的离子半径比Zn更大,而平均压缩率显示出最小的变化。然而,人们发现,在较高掺杂剂含量下,V 的掺入可使 ZnS 软化超过 15%,从而增加柔量和各向异性。稳定的铁磁自旋耦合出现,由主载流子介导,而不沉淀 V 第二相。该能带结构随着 V-3d 和 S-3p 杂化效应引起的自旋简并性和优先自旋能带金属性的提升而发展出半金属电子特性。这导致费米能级处 100% 自旋极化。基于由最大局域 Wannier 函数构建的紧束缚哈密顿量的蒙特卡罗模拟能够准确计算 V 自旋之间的交换相互作用,成功地再现了磁相变行为。结果证实,25% V 时居里温度超过 300 K,这归因于短程铁磁耦合相对于反铁磁相互作用的显着增强。集成计算方法为掺杂 V 纤锌矿 ZnS 稀磁半导体中载流子介导的铁磁性的微观起源提供了基本见解。系统的第一性原理计算和统计力学模型建立了清晰的结构-性能关系,将 V 掺杂构型和浓度与 ZnS 中的机械响应、稳定性增强、电子结构修饰、自旋极化和磁有序温度联系起来。研究结果提出了设计途径和预测能力,以指导自旋电子技术的新兴高性能磁性材料的实验工作。


























 京公网安备 11010802027423号
京公网安备 11010802027423号