Advanced Synthesis & Catalysis ( IF 5.4 ) Pub Date : 2024-03-11 , DOI: 10.1002/adsc.202400146 Chengtao Zhao 1 , Tatiana Besset 2 , PHILIPPE JUBAULT 3
|
In the field of organic synthesis for the design of new bioactive molecules, the fluorine atom and more generally fluorinated groups have attracted significant attention from the scientific community because of the abilities of these groups to modify the physico-chemical properties of a target molecule.1 The main consequences of the installation of a fluorinated group onto a molecule are the modulation of numerous properties such as lipophilicity, metabolic stability, and the acidity/basicity of neighboring functions.2 As a consequence, very important breakthroughs have been reported for the synthesis of numerous fluorine-containing molecules.3 Moreover, there is a growing interest in the incorporation of a CF2H group because this specific fluorinated group can be used as a bioisoster of a methyl or a thiol group for example.4 In addition, the α,α-difluoromethylcyclopropane motif is present in several bioactive and marketed drugs such as Volaxiprevir and Glecaprevir for the treatment of Hepatitis C5 and more recently Lenacapavir as HIV-1 capsid inhibitor.6 Quite surprisingly and despite the important potential of this cyclopropyl motif, the access to difluoromethylated cyclopropanes remains scarce.7 Since pioneering and very important reports from Savins, Hao, Hanamoto, Leadbeater, Lin, Xiao and Koenigs for the synthesis of such racemic CF2H-cyclopropanes,8 only a few original approaches have emerged recently. For example, Bi and co-workers9 developed a bench-stable difluoroacetaldehyde N-triftosylhydrazone (DFHZ-Tfs) as a CF2HCHN2 diazo surrogate, which exhibits a remarkable carbene reactivity in the FeIII-catalyzed cyclopropanation (Scheme 1, eq. 1). In 2022, Gilmour and co-workers10 reported a fluorinative ring contraction of aryl-substituted cyclobutene derivatives under a I(I)/I(III) catalysis leading to the cis-α,α-difluoromethylcyclopropanes in good yields and dr (Scheme 1, eq. 2). Finally, in 2023, Ma's group11 reported the synthesis of CF2SO2Ph cyclopropane-dicarbonitriles via [2+1] cycloaddition of difluoro-based diazoethanes with alkylidene malonodinitriles in very good yields (Scheme 1, eq. 3).

Recent racemic strategies to access cyclopropanes bearing a CF2H or a CF2-substituted group. FeTPPCl: 5,10,15,20-Tetraphenyl-21H,23H-porphine iron (III) chloride.
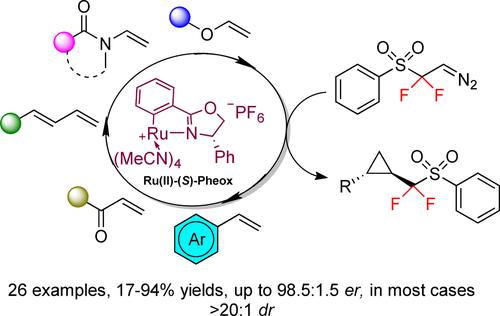
Regarding the access to chiral difluoromethylated cyclopropanes, no asymmetric process has been reported until 2017. Indeed, using the dirhodium paddlewheel complex Rh2((S)-BTPCP)4, our group reported the cyclopropanation of difluoromethylated styrenes12 in the presence α-aryl diazo acetates and α-nitro diazoketones to access functionalized tetrasubstituted CF2H-containing cyclopropanes in high dr and ee’s (Scheme 2, eq. 1). In 2019, Marek, Zhang, Ma and co-workers13 reported an efficient RhII-catalyzed enantioselective cyclopropenation reaction of internal alkynes with PhSO2CF2CHN2 diazo reagent leading to the expected chiral cyclopropenes with very good yields and stereoselectivities. For a single example, the corresponding trisubstituted PhSO2CF2-cyclopropane was obtained after hydrogenation of the corresponding cyclopropene (Scheme 2, eq. 2). In 2020, the group of Zhang reported a very efficient strategy for the stereoselective functionalization of racemic cyclopropylzinc reagents with a broad range of gem-difluoropropargyl bromides leading to the expected cis disubstituted CF2-cyclopropanes with excellent levels of enantioselectivity (Scheme 2, eq. 3).14 One year later, Fasan and our group reported an enantioselective biocatalytic cyclopropanation of α-difluoromethylated olefins by ethyl diazoacetate using two engineered myoglobin enzymes.15 The trisubstituted difluoromethylated cyclopropanes were obtained in good and excellent enantioselectivities (Scheme 2, eq. 4). In 2023, Bi and co-workers16 reported the synthesis of chiral CF2CO2Et-substituted cyclopropanes through enantioselective [2+1] cyclopropanation of alkenes and DFHZ-Tfs under rhodium catalysis. The expected tetrasubstituted cyclopropyl motifs were obtained in high yields and enantioselectivity (Scheme 2, eq. 5). Finally, Wu, Zhu and co-workers17 reported very recently a two-steps access to enantioenriched tetra-substituted CF2H cyclopropanes via an Rh(II)-catalyzed intramolecular cycloisomerization of CF2H-substituted enynes followed by ozonolysis. For six examples, the expected CF2H cyclopropanes were in good to very yields and excellent enantioselectivities (Scheme 2, eq. 6). Despite these important and recent contributions, a general and efficient approach for the stereoselective synthesis of trans-1,2 disubstituted cyclopropanes bearing a CF2FG (FG=functional group) or CF2H group remained unknown. Inspired by the previous works using the appealing CF2SO2Ph moiety18 as an interesting fluorinated group and a potent synthetic platform to access the important CF2H residue, we surmise that the development of a catalytic enantioselective methodology enabling the synthesis of CF2SO2Ph-containing cyclopropanes would provide straightforward access to original high-value added fluorinated scaffolds. As part of our research program dedicated to the development of original methodologies to access chiral cyclopropanes19 in particularly fluorinated ones, we report, herein, the ruthenium-catalyzed asymmetric cyclopropanation of olefins with PhSO2CF2CHN2 diazo reagent in very high diasteroselectivities and enantioselectivities.

Catalytic enantioselective strategies to access cyclopropanes bearing a CF2H or a CF2-substituted group. Rh2((S)-BTPCP)4: Dirhodium tetrakis[(S)-1-(4-bromophenyl)-2,2- diphenylcyclopropane carboxylate]. Rh2((S)PTAD)4:Tetrakis[(S)-(1-adamantyl)-(N-phthalimido)acetate] dirhodium(II). TFT=Tri-fluorotoluene.
At the outset of this project, we investigated the reaction between styrene 1 a and the PhSO2CF2CHN2 diazo reagent20 2 as the carbene precursor in the presence of 2 mol% of a chiral ruthenium catalyst (Table 1). First, Ru(II)(S)-Pheox I21 was tested in CH2Cl2 at room temperature in the presence of 1 equiv. of 2 and an excess of styrene 1 a (5 equiv., entry 1). As the result, the desired cyclopropane 3 a substituted with a phenylsulfonyldifluoromethyl group was obtained in a good NMR yield as a >20:1 diastereoisomeric ratio with an excellent enantiomeric ratio (entry 1). Inverting the ratio between 1 a and 2, we observed a decrease in the NMR yield (for 1 equiv. of 1 a and 1.5 equiv. of 2, 37% NMR yield). We then screened the influence of the solvent on the outcome of the cyclopropanation. Toluene, THF or diethyl ether led to lower NMR yields but with almost the same stereoselectivity (entries 2–4). The use of DCE led to the best NMR yield (70%) with excellent stereoselectivity (>20:1 d.r. and 95:5 e.r., entry 5). We next tested another Ru-(II) catalyst (entry 6) and two rhodium(II) complexes Rh2((S)-PhTPCP)4 and Rh2((S)-TCPTTL)4 but these chiral catalysts led to lower NMR yields and modest stereoselectivities in the case of rhodium(II) catalysts (entries 7, 8). Increasing the amount of Ru(II)(S)-Pheox I to 5 mol% led to a better NMR yield and the expected cyclopropane 3 a was isolated in 74% yield. Decreasing the temperature to 0 °C or −10 °C had a negative impact on the outcome of the reaction lower NMR yields were observed (59% and 37%, respectively). Finally, the optimal conditions were delineated to achieve the highest yield, diastereo- and enantioselectivity, using Ru(II)(S)-Pheox I (5 mol%) at room temperature in dichloroethane with 5 equiv. of styrene 1 a and 1 equiv. of 2 (entry 9). Worth mentioning that the (R,R)-absolute configuration was unambiguously determined by the X-ray crystallographic analysis on 3 a.22
|
|||||
Entry |
[catalyst] |
solvent |
NMR yield [%] b) |
d.r.[b] |
e.r.[c] |
|---|---|---|---|---|---|
1 |
Ru(II)(S)-Pheox I |
CH2Cl2 |
49 |
>20:1 |
95:5 |
2 |
Ru(II)(S)-Pheox I |
Tol |
24 |
>20:1 |
90:10 |
3 |
Ru(II)(S)-Pheox I |
THF |
37 |
>20:1 |
93:7 |
4 |
Ru(II)(S)-Pheox I |
Et2O |
16 |
>20:1 |
92:8 |
5 |
Ru(II)(S)-Pheox I |
DCE |
70 |
>20:1 |
95:5 |
6 |
Ru(II)(S)-Pheox II |
DCE |
33 |
>20:1 |
91:9 |
7 |
Rh2((S)-PhTPCP)4 |
DCE |
28 |
38:62 |
78:22 |
8 |
Rh2(S-TCPTTL)4 |
DCE |
57 |
30:70 |
80:20 |
9[d] |
Ru(II)((S)-Pheox I |
DCE |
83 (74)[e] |
>20:1 |
95:5 |
|
|||||


- [a] To a mixture of styrene 1 a (5 equiv.) and catalyst (2 mol%) in a solvent, a solution of 2 (1 equiv.) in a solvent was slowly added over 1 h via a syringe pump at room temperature. [b] NMR yield determined by 1H NMR analysis of the crude reaction mixture using CH2Br2 as an internal standard. [c] Determined by HPLC analysis on a chiral stationary phase. [d] 5 mol% of catalyst was used. [e] Isolated yield. Tol=toluene.
With these optimized reaction conditions in hand, a variety of styrene derivatives was investigated for this asymmetric cyclopropanation using 2 as a carbene partner (Scheme 3). The effect of the methoxy group at the para-position of the aryl group was examined using para-methoxy styrene and the desired cyclopropane 3 b was afforded in high yield (75%), excellent diastereoselectivity (>20:1 d.r.) and high enantiomeric ratio (94:6).

Cyclopropanation of 1 a with diazo 2. To a mixture of styrene 1 (5 equiv.) and Ru(II)(S)-Pheox I (5 mol%) in DCE (0.5 mL), a solution of 2 (1 equiv.) in a DCE (1 mL) was slowly added over 1 h via a syringe pump at room temperature. a) Isolated yield. b) The diastereomeric ratio was determined by 1H NMR analysis of the crude reaction mixture. c) The enantiomeric ratio was determined by HPLC analysis on a chiral stationary phase.
The reaction was tolerant to the presence of a methyl group at different positions (para, meta, ortho) on the phenyl ring as the expected cyclopropanes 3 c–e were obtained in good isolated yields (61–70%) and excellent stereoselectivities in each case. The presence of halogen atom (chlorine, bromine, iodine) at the para, meta or ortho position on the aryl substituted alkene did not impact the efficiency of the cyclopropanation, giving the desired products 3 f–j in good to very good isolated yields (57–78%), excellent diastereoselectivity and very good enantiomeric ratios. However, when 2,6-dichlorostyrene was used, the corresponding cyclopropane 3 k was obtained in low yield (17%) and the er decreased to 83:17. Regarding the substitution of the phenyl ring at the para position, the cyclopropanation occurred very efficiently with a phenyl group or a bis-pinacolborane substituent leading to the desired products 3 l–m in good yields and stereoselectivites. On the contrary, the presence of an electron-withdrawing group (CF3, NO2) at the para or meta position on the phenyl ring led to an important decrease of the isolated yields (26–38%) for the expected cyclopropanes 3 n–p but the dr and the er remain high, expect in the case of 3 p (80:20 dr). Interestingly, the cyclopropanation process proceeded smoothly with other aromatic alkenes such as 2-vinyl naphthalene, 9-vinyl anthracene, vinyl ferrocene and 2-vinyl benzofuran leading to the expected cyclopropanes 3 q–r in good to excellent yields (65–92%) and again the stereoselectivities remained very high for these four examples (dr >20:1 and er from 95:5 to 98:2). However, our reaction showcased some limitations as when 3-phenylprop-1-ene or 4-vinylpyridine was used, the expected cyclopropane was not detected.
To further broaden the portfolio of enantioenriched trans 1,2-phenylsulfonyldifluoromethylated substituted cyclopropanes 3 and after exploring the scope of aromatic styrene derivatives, we investigated the cyclopropanation of other classes of alkenes (Scheme 4).

Scope of the cyclopropanation of various alkenes 1 with reagent 2 and reluctant substrates. To a mixture of alkene 1 (5 equiv.) and catalyst (5 mol%) in DCE (0.5 mL), a solution of 2 (1 equiv.) in a DCE (1 mL) was slowly added over 1 h via a syringe pump at room temperature. a) Isolated yield. b) The diastereomeric ratio was determined by 1H NMR analysis of the crude reaction mixture. c) The enantiomeric ratio was determined by HPLC analysis on a chiral stationary phase.
Unsaturated ketone 1 u was readily converted into the corresponding cyclopropane 3 u in good yield with excellent diastereo- and enantioselectivity. Benzyl vinylcarbamate 1 v led very efficiently to the desired product 3 v in a very good yield (84%) in a 60:40 mixture of separable diastereoisomers. For the trans product, the er remained excellent (97% ee) whereas an important decrease was observed for the cis one (67:33). Interestingly, the use of N-vinylphthalimide 1 w in the cyclopropanation reaction led to very high stereoselectivities for cyclopropane 3 w but we observed a moderate isolated yield (24%) probably because of steric hindrance. Trans-1-phenyl-1,3-butadiene 1 x was then tested and pleasingly the reaction proceeded smoothly and the corresponding product 3 x was isolated in very good yield (85%), excellent dr and er (>20:1 and 96.5:3.5 respectively). Finally, vinyl acetate 1 y and phenyl vinyl ether 1 z were engaged in the cyclopropanation reaction leading to the expected cyclopropanes 3 y and 3 z with very good stereoselectivites in both cases and moderate yield for 3 y (45%) and an excellent one for 3 z (94%). However, our reaction showcased some limitations. Indeed, the cyclopropanation reaction using diphenylethene 1 aa, vinylsilane 1 ab, unsaturated amide 1 ac or ethyl acrylate 1 ad was inefficient as no trace of the expected cyclopropane was detected by 1H NMR on the crude mixture.
It is important to note that a scale-up of the reaction (Scheme 5) with 1 a, 1 m and 1 u on 3 mmol (4 mmol for 1 m) was carried out. The expected cyclopropanes 3 a, 3 m and 3 u were isolated in 75%, 70% and 55% isolated yields, respectively, with the same excellent diastereoselectivities and enantiomeric ratios demonstrating the robustness of the process.

Scale-up of the cyclopropanation reaction.
The versatility of the resulting cyclopropanes 3 can be illustrated by their further post-functionalization (Scheme 6). First, the addition of phenyl magnesium bromide on cyclopropane 3 u afforded the corresponding tertiary alcohol 4 a in a very good 82% yield whereas a slight erosion of the enantiomeric ratio was observed (95:5 for 4 a compared to 98:2 for 3 u). Then, cyclopropane 3 u was efficiently reduced to the corresponding secondary alcohol 4 b in 86% as a 78:28 mixture of diastereoisomers. In addition, cyclopropane 3 u can be converted into 4 c through a Wittig reaction in the presence of phosphonium salt Ph3P+CH3, I− and n-BuLi as a base in 89% yield and 98:2 er. Then, the PhSO2CF2 unit was smoothly converted to unprecedented and high value-added trans 1,2-disubstituted difluoromethylated cyclopropanes. At first, reaction conditions used by Ma and co-workers for the reduction of PhSO2CF2-containing cyclopropenes were tested.11 However, when applied to the cyclopropanes 3, only traces of the expected products were observed. Then, the reduction of cyclopropane 4 c was carried out using 3% Na−Hg amalgam,23 leading to the corresponding CF2H-containing cyclopropane 4 d in a very good yield (74%) and without any erosion of the er (97:3). The same protocol was efficiently applied to cyclopropanes 3 a and 3 z affording the expected CF2H-containing cyclopropanes 4 e and 4 f in 88 and 83% yields, respectively. In both cases, the enantiomeric ratios remained excellent.

Post-functionalization of cyclopropanes 3 and access to high added-value CF2H-containing cyclopropanes.
In conclusion, we disclosed the catalytic enantioselective synthesis of 1,2-trans-disubstituted PhSO2CF2-containing cyclopropanes from different classes of alkenes and the PhSO2CF2CHN2 diazo derivative as a CF2HCHN2 surrogate. The use of a chiral catalyst, i. e. Ru(II)-Pheox I, furnished the expected cyclopropanes (26 examples) in high yields and stereoselectivities. This reaction manifold allowed, through the reduction of the benzene sulfonyl group, the synthesis of the high value-added trans 1,2-disubtituted difluoromethylated cyclopropanes.
Experimental Section
An oven-dried 10 mL reaction tube contained a stir bar, catalyst Ru(II)(S)-Pheox I (0.005 mmol, 0.05 equiv.) which was stored in a glovebox was introduced to the reaction tube in this glovebox, followed by alkene 1 (0.5 mmol, 5.0 equiv.) under argon atmosphere. Fresh distilled DCE 0.5 mL was added via a syringe. Diazo compound 2 (23.2 mg, 0.1 mmol, 1.0 equiv.) was dissolved in 1.0 mL fresh distilled DCE and added to the former solution with 1.0 mL/h rate by use of microinjection pump. When the addition finished, the completion of reaction was detected by TLC. The NMR yield and diastereomeric ratio were detected with 1H NMR at the end of this reaction (dibromomethane as internal standard). The reaction mixture was evaporated under reduce pressure and purified by silica gel chromatography leading to the corresponding enantiopure cyclopropanes 3.
Acknowledgments
This work was partially supported by Normandie Université (NU), the Région Normandie, the Centre National de la Recherche Scientifique (CNRS), Université de Rouen Normandie (URN), INSA Rouen Normandie, Labex SynOrg (ANR-11-LABX-0029), the graduate school for research XL–Chem (ANR-18-EURE-0020 XL CHEM) and Innovation Chimie Carnot (I2 C). T.B. thank the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement no. 758710). C. Z. thanks the China Scholarship Council for a doctoral fellowship.
中文翻译:
钌(II)催化对映选择性合成含1,2-反式二取代PhSO2CF2的环丙烷
在用于设计新生物活性分子的有机合成领域中,氟原子和更普遍的氟化基团由于这些基团能够改变目标分子的物理化学性质而引起了科学界的极大关注。1在分子上安装氟化基团的主要后果是调节许多特性,例如亲脂性、代谢稳定性以及邻近功能的酸度/碱度。2因此,许多含氟分子的合成取得了非常重要的突破。3此外,人们对掺入 CF 2 H 基团越来越感兴趣,因为这种特定的氟化基团可以用作例如甲基或硫醇基团的生物等排体。4此外,α,α-二氟甲基环丙烷基序存在于多种生物活性药物和市售药物中,例如用于治疗丙型肝炎5的 Volaxiprevir 和 Glecaprevir以及最近作为 HIV-1 衣壳抑制剂的 Lenacapavir。6令人惊讶的是,尽管这种环丙基基序具有重要的潜力,但获得二氟甲基化环丙烷的途径仍然很少。7自从 Savins、Hao、Hanamoto、Leadbeater、Lin、Xiao 和 Koenigs 提出合成此类外消旋 CF 2 H-环丙烷的开创性且非常重要的报告以来,8最近仅出现了一些原始方法。例如,Bi 和同事9开发了一种实验室稳定的二氟乙醛N -三甲苯基腙 (DFHZ-Tfs) 作为 CF 2 HCHN 2重氮替代物,它在 Fe III催化的环丙烷化反应中表现出显着的卡宾反应活性(方案 1,eq) . 1). 2022 年,Gilmour 和同事10报道了芳基取代的环丁烯衍生物在 I(I)/I(III) 催化下的氟化环收缩,产生了顺式-α,α-二氟甲基环丙烷,且产率良好(方案 1) ,等式2)。最后,在 2023 年,Ma 的第 11组报道了通过二氟基重氮乙烷与亚烷基丙二腈的 [2+1] 环加成反应合成了 CF 2 SO 2 Ph 环丙烷二甲腈,收率非常高(方案 1,方程式 3)。
最近获得带有CF 2 H或CF 2取代基团的环丙烷的外消旋策略。 FeTPPCl:5,10,15,20-四苯基-21 H ,23 H-卟吩氯化铁(III)。
关于获得手性二氟甲基化环丙烷,直到 2017 年才报道不对称过程。事实上,使用二铑叶轮络合物 Rh 2 (( S) -BTPCP) 4 ,我们的小组报道了在 α-芳基存在下二氟甲基化苯乙烯12的环丙烷化反应重氮乙酸酯和 α-硝基重氮酮在高dr和ee下得到官能化的四取代 CF 2 H 含环丙烷(方案 2,eq. 1)。 2019年,Marek、Zhang、Ma和同事13报道了一种高效的Rh II催化的内炔与PhSO 2 CF 2 CHN 2重氮试剂的对映选择性环丙烯化反应,产生了具有非常好的产率和立体选择性的预期手性环丙烯。就单个实例而言,在相应的环丙烯氢化后获得相应的三取代的PhSO 2 CF 2 -环丙烷(方案2,eq.2)。 2020年,张教授团队报道了一种非常有效的策略,可以用多种偕二氟炔丙基溴对外消旋环丙基锌试剂进行立体选择性官能化,从而产生具有优异对映选择性水平的顺式二取代CF 2 -环丙烷(方案2,eq.1)。 3)。14一年后,Fasan 和我们的小组报道了使用两种工程化肌红蛋白酶,通过重氮乙酸乙酯对 α-二氟甲基化烯烃进行对映选择性生物催化环丙烷化反应。15获得的三取代二氟甲基化环丙烷具有良好且优异的对映选择性(方案 2,eq. 4)。 2023年,Bi和同事16报道了在铑催化下通过烯烃和DFHZ-Tfs的对映选择性[2+1]环丙烷化合成了手性CF 2 CO 2 Et取代的环丙烷。以高产率和对映选择性获得了预期的四取代环丙基基序(方案 2,eq. 5)。最后,Wu、Zhu 和同事17最近报道了通过Rh(II) 催化的CF 2 H取代烯炔的分子内环异构化,然后进行臭氧分解,分两步获得对映体富集的四取代CF 2 H 环丙烷。对于六个示例,预期 CF 2H 环丙烷具有良好至非常高的产率和优异的对映选择性(方案 2,方程式 6)。尽管有这些重要且最新的贡献,但是用于立体选择性合成带有CF 2 FG(FG=官能团)或CF 2 H基团的反式-1,2二取代环丙烷的通用且有效的方法仍然未知。受到之前使用引人注目的 CF 2 SO 2 Ph 部分18作为有趣的氟化基团和获取重要 CF 2 H 残基的有效合成平台的工作的启发,我们推测催化对映选择性方法的开发能够合成 CF 2含SO 2 Ph的环丙烷将提供直接获得原始高附加值氟化支架的途径。作为我们致力于开发原始方法以获取手性环丙烷19特别是氟化环丙烷的研究计划的一部分,我们在此报告了钌催化的烯烃与 PhSO 2 CF 2 CHN 2重氮试剂在非常高的非对映选择性下的不对称环丙烷化反应,并且对映选择性。
获得带有CF 2 H或CF 2取代基团的环丙烷的催化对映选择性策略。 Rh 2 (( S) -BTPCP) 4:四[( S )-1-(4-溴苯基)-2,2-二苯基环丙烷甲酸二铑]。 Rh 2 (( S )PTAD) 4:四[( S )-(1-金刚烷基)-( N-邻苯二甲酰亚氨基)乙酸]二铑(II)。 TFT=三氟甲苯。
在该项目开始时,我们研究了苯乙烯1 a与作为卡宾前体的 PhSO 2 CF 2 CHN 2重氮试剂20 2在 2 mol% 手性钌催化剂存在下的反应(表 1)。首先,在室温下、在1当量存在下,在CH 2 Cl 2中测试Ru(II)( S) -Pheox I 21 。 2和过量的苯乙烯1 a(5 当量,条目 1)。结果,以良好的NMR产率获得了所需的被苯磺酰基二氟甲基取代的环丙烷3a,其非对映异构体比率>20:1,具有优异的对映异构体比率(条目1)。反转1 a和2之间的比率,我们观察到 NMR 产率下降(对于 1 当量1 a和 1.5 当量2,NMR 产率为 37%)。然后我们筛选了溶剂对环丙烷化结果的影响。甲苯、THF 或乙醚导致 NMR 产率较低,但立体选择性几乎相同(条目 2-4)。使用 DCE 可获得最佳 NMR 产率 (70%) 和出色的立体选择性(>20:1 dr 和 95:5 er,条目 5)。接下来,我们测试了另一种 Ru-(II) 催化剂(条目 6)和两种铑 (II) 配合物 Rh 2 (( S)- PhTPCP) 4和 Rh 2 (( S)- TCPTTL) 4,但这些手性催化剂导致 NMR 较低在铑 (II) 催化剂的情况下,产率和适度的立体选择性(条目 7、8)。将 Ru(II)( S)-Pheox I的量增加至 5 mol% 会带来更好的 NMR 产率,并且以 74% 的产率分离出预期的环丙烷3a 。将温度降低至 0 °C 或 -10 °C 对反应结果产生负面影响,观察到较低的 NMR 产率(分别为 59% 和 37%)。最后,在室温下使用 Ru(II)( S)-Pheox I ( 5 mol%) 在二氯乙烷中使用 5 当量的溶液,确定了实现最高收率、非对映选择性和对映选择性的最佳条件。苯乙烯1a和1当量。共2 个(条目 9)。值得一提的是,( R,R )-绝对构型是通过对3a的 X 射线晶体学分析明确确定的。22
|
|||||
入口 |
[催化剂] |
溶剂 |
核磁共振 产率 [%] b) |
博士[b] |
呃[c] |
|---|---|---|---|---|---|
1 |
Ru(II)( S)- Pheox I |
氯甲烷2 |
49 |
>20:1 |
95:5 |
2 |
Ru(II)( S)- Pheox I |
托尔 |
24 |
>20:1 |
90:10 |
3 |
Ru(II)( S)- Pheox I |
四氢呋喃 |
37 |
>20:1 |
93:7 |
4 |
Ru(II)( S)- Pheox I |
乙醚2O |
16 |
>20:1 |
92:8 |
5 |
Ru(II)( S)- Pheox I |
大连商品交易所 |
70 |
>20:1 |
95:5 |
6 |
Ru(II)( S) -Pheox II |
大连商品交易所 |
33 |
>20:1 |
91:9 |
7 |
Rh 2 (( S) -PhTPCP) 4 |
大连商品交易所 |
28 |
38:62 |
78:22 |
8 |
Rh 2 ( S -TCPTTL) 4 |
大连商品交易所 |
57 |
30:70 |
80:20 |
9 [d] |
Ru(II)(( S)- Pheox I |
大连商品交易所 |
83 (74) [英] |
>20:1 |
95:5 |
|
|||||
- [a]在室温下,通过注射泵在1小时内向苯乙烯1a(5当量)和催化剂(2mol%)在溶剂中的混合物中缓慢添加2 (1当量)在溶剂中的溶液。温度。[b] NMR产率通过使用CH 2 Br 2作为内标对粗反应混合物进行1 H NMR分析来确定。 [c]通过手性固定相的 HPLC 分析测定。[d]使用5mol%的催化剂。[e]分离产量。 Tol=甲苯。
掌握这些优化的反应条件后,使用2作为卡宾伴侣,研究了各种苯乙烯衍生物的不对称环丙烷化反应(方案 3)。使用对甲氧基苯乙烯检查了芳基对位上的甲氧基的影响,并以高产率(75%)、优异的非对映选择性(>20:1 dr)和高对映体得到了所需的环丙烷3b比例(94:6)。
1 a与重氮2的环丙烷化。向苯乙烯1(5 当量)和 Ru(II)( S)-Pheox I ( 5 mol%) 在 DCE (0.5 mL) 中的混合物中加入2(1 当量)在 DCE (1 mL) 中的溶液在室温下通过注射泵在1小时内缓慢添加。 a) 孤立产量。 b)通过粗反应混合物的1 H NMR分析确定非对映体比率。 c) 通过手性固定相上的 HPLC 分析确定对映体比例。
该反应能够耐受苯环上不同位置(对位、间位、邻位)甲基的存在,因为预期的环丙烷3 c – e均以良好的分离产率 (61–70%) 获得,并且每个反应都具有出色的立体选择性。案件。芳基取代烯烃的对位、间位或邻位上卤素原子(氯、溴、碘)的存在不会影响环丙烷化的效率,从而以良好至非常好的分离产率得到所需的产物3 f – j( 57–78%),优异的非对映选择性和非常好的对映体比例。然而,当使用2,6-二氯苯乙烯时,得到相应的环丙烷3k,产率较低(17%),er 降低至83:17。关于苯环在对位的取代,环丙烷化与苯基或双频哪醇硼烷取代基发生非常有效,从而以良好的产率和立体选择性产生所需的产物3 l – m 。相反,苯环上对位或间位吸电子基团(CF 3、NO 2)的存在导致预期环丙烷3 n的分离收率显着降低(26-38%) - p但 dr 和 er 仍然很高,预计3 p (80:20 dr)的情况。有趣的是,与其他芳香族烯烃(例如 2-乙烯基萘、9-乙烯基蒽、乙烯基二茂铁和 2-乙烯基苯并呋喃)的环丙烷化过程顺利进行,产生了预期的环丙烷3 q – r,收率良好至优异(65-92%)这四个例子的立体选择性仍然非常高(dr >20:1 和 er 从 95:5 到 98:2)。然而,我们的反应显示出一些局限性,因为当使用 3-苯基丙-1-烯或 4-乙烯基吡啶时,未检测到预期的环丙烷。
为了进一步拓宽对映体富集的反式1,2-苯磺酰基二氟甲基化取代环丙烷3的产品组合,并在探索芳香族苯乙烯衍生物的范围后,我们研究了其他类别烯烃的环丙烷化(方案 4)。
使用试剂2和惰性底物对各种烯烃1进行环丙烷化的范围。向烯烃1 (5 当量)和催化剂(5 mol%)在 DCE(0.5 mL)中的混合物中,通过注射器在 1 小时内缓慢添加2 (1 当量)在 DCE(1 mL)中的溶液室温下泵。 a) 孤立产量。 b)通过粗反应混合物的1 H NMR分析确定非对映体比率。 c) 通过手性固定相上的 HPLC 分析确定对映体比例。
不饱和酮1 u很容易转化为相应的环丙烷3 u,收率良好,具有优异的非对映选择性和对映选择性。乙烯基氨基甲酸苄酯1 v在可分离的非对映异构体的 60:40 混合物中以非常好的收率 (84%)非常有效地生成所需产物3 v 。对于反式产品,er 仍然出色 (97% ee),而顺式产品则观察到显着下降( 67:33)。有趣的是,在环丙烷化反应中使用N-乙烯基邻苯二甲酰亚胺1 w导致环丙烷3 w具有非常高的立体选择性,但我们观察到中等的分离产率(24%),可能是由于空间位阻。然后测试反式-1-苯基-1,3-丁二烯1 x,令人高兴的是,反应顺利进行,相应的产物3 x以非常好的产率(85%)、优异的 dr 和 er(>20:1 和 96.5)分离出来:分别为3.5)。最后,乙酸乙烯酯1y和苯基乙烯基醚1z进行环丙烷化反应,生成预期的环丙烷3y和3z,在两种情况下都具有非常好的立体选择性,并且3y 的产率适中(45%),对于3z(94%)。然而,我们的反应显示出一些局限性。事实上,使用二苯乙烯1 aa、乙烯基硅烷1 ab、不饱和酰胺1 ac或丙烯酸乙酯1 ad 的环丙烷化反应效率低下,因为通过1 H NMR在粗混合物上没有检测到痕量的预期环丙烷。
值得注意的是,使用1a、1m和1u在 3mmol 上进行了反应(方案 5)的放大( 1m为 4mmol)。预期的环丙烷3a、3m和3u分别以 75%、70% 和 55% 的分离产率分离,具有同样出色的非对映选择性和对映体比率,证明了该方法的稳健性。
环丙烷化反应的放大。
所得环丙烷3的多功能性可以通过其进一步的后官能化来说明(方案 6)。首先,在环丙烷3 u上添加苯基溴化镁,得到相应的叔醇4 a,收率非常好,为 82%,但观察到对映体比例略有下降(4 a为 95:5,3 a为 98:2 )。你)。然后,环丙烷3u被有效还原为86%的相应仲醇4b,为78:28的非对映异构体混合物。此外,在鏻盐Ph 3 P + CH 3、I -和n -BuLi作为碱存在下,通过维蒂希反应可以将环丙烷3 u转化为4 c ,产率89%,er 98:2。然后,PhSO 2 CF 2装置顺利转化为前所未有的高附加值反式1,2-二取代二氟甲基化环丙烷。首先,测试了Ma及其同事用于还原含PhSO 2 CF 2的环丙烯的反应条件。 11然而,当应用于环丙烷3时,仅观察到了预期产物的痕迹。然后,使用 3% Na−Hg 汞齐进行环丙烷4 c的还原, 23得到相应的含 CF 2 H 的环丙烷4 d,产率非常好(74%),并且没有任何侵蚀(97 :3)。将相同的方案有效地应用于环丙烷3a和3z,分别以88%和83%的产率提供预期的含CF 2 H的环丙烷4e和4f 。在这两种情况下,对映体比率仍然非常好。
环丙烷3的后官能化并获得高附加值的含CF 2 H环丙烷。
总之,我们公开了从不同类别的烯烃催化对映选择性合成含1,2-反式二取代的PhSO 2 CF 2环丙烷和作为CF 2 HCHN 2替代物的PhSO 2 CF 2 CHN 2重氮衍生物。手性催化剂的使用,即。 e . Ru(II)-Pheox I以高产率和立体选择性提供了预期的环丙烷(26 个实例)。该反应流形通过苯磺酰基的还原,合成高附加值反式1,2-二取代二氟甲基化环丙烷。
实验部分
将烘箱干燥的装有搅拌棒的10mL反应管,将储存在手套箱中的催化剂Ru(II)( S)-Pheox I(0.005mmol,0.05当量)引入到该手套箱中的反应管中,然后氩气气氛下的烯烃1(0.5 mmol,5.0 当量)。通过注射器添加新鲜蒸馏的 DCE 0.5 mL。将重氮化合物2(23.2 mg,0.1 mmol,1.0当量)溶解在1.0 mL新鲜蒸馏的DCE中,并使用微注射泵以1.0 mL/h的速率添加到前溶液中。添加完成后,通过TLC检测反应完成。反应结束时用1 H NMR检测 NMR 产率和非对映异构体比例(二溴甲烷作为内标)。将反应混合物减压蒸发并通过硅胶色谱纯化,得到相应的对映体纯环丙烷3。
致谢
这项工作得到了诺曼底大学 (NU)、诺曼底地区、国家科学研究中心 (CNRS)、鲁昂诺曼底大学 (URN)、INSA Rouen Normandie、Labex SynOrg (ANR-11-LABX-0029) 的部分支持,研究 XL–Chem (ANR-18-EURE-0020 XL CHEM) 和创新 Chimie Carnot (I2 C) 的研究生院。 TB 感谢欧洲研究理事会 (ERC) 根据欧盟地平线 2020 研究和创新计划(拨款协议编号 758710)。 CZ感谢国家留学基金委提供的博士奖学金。


























 京公网安备 11010802027423号
京公网安备 11010802027423号