GeroScience ( IF 5.6 ) Pub Date : 2024-01-25 , DOI: 10.1007/s11357-023-01059-y Roman Abrosimov , Marius W. Baeken , Samuel Hauf , Ilka Wittig , Parvana Hajieva , Carmen E. Perrone , Bernd Moosmann
|
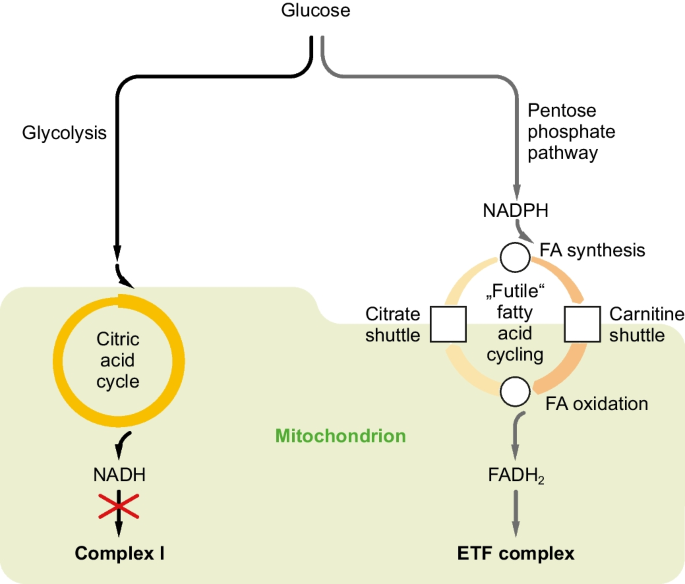
Inhibition of mitochondrial complex I (NADH dehydrogenase) is the primary mechanism of the antidiabetic drug metformin and various unrelated natural toxins. Complex I inhibition can also be induced by antidiabetic PPAR agonists, and it is elicited by methionine restriction, a nutritional intervention causing resistance to diabetes and obesity. Still, a comprehensible explanation to why complex I inhibition exerts antidiabetic properties and engenders metabolic inefficiency is missing. To evaluate this issue, we have systematically reanalyzed published transcriptomic datasets from MPP-treated neurons, metformin-treated hepatocytes, and methionine-restricted rats. We found that pathways leading to NADPH formation were widely induced, together with anabolic fatty acid biosynthesis, the latter appearing highly paradoxical in a state of mitochondrial impairment. However, concomitant induction of catabolic fatty acid oxidation indicated that complex I inhibition created a “futile” cycle of fatty acid synthesis and degradation, which was anatomically distributed between adipose tissue and liver in vivo. Cofactor balance analysis unveiled that such cycling would indeed be energetically futile (-3 ATP per acetyl-CoA), though it would not be redox-futile, as it would convert NADPH into respirable FADH2 without any net production of NADH. We conclude that inhibition of NADH dehydrogenase leads to a metabolic shift from glycolysis and the citric acid cycle (both generating NADH) towards the pentose phosphate pathway, whose product NADPH is translated 1:1 into FADH2 by fatty acid cycling. The diabetes-resistant phenotype following hepatic and intestinal complex I inhibition is attributed to FGF21- and GDF15-dependent fat hunger signaling, which remodels adipose tissue into a glucose-metabolizing organ.
中文翻译:
线粒体复合物 I 抑制通过连续的 NADPH 形成、“无效”脂肪酸循环和 FADH2 氧化触发 NAD+ 独立的葡萄糖氧化
抑制线粒体复合物 I(NADH 脱氢酶)是抗糖尿病药物二甲双胍和各种不相关的天然毒素的主要机制。复合物 I 抑制也可以由抗糖尿病 PPAR 激动剂诱导,并且是由蛋氨酸限制引起的,蛋氨酸限制是一种导致糖尿病和肥胖抵抗的营养干预措施。然而,对于复合物 I 抑制为何发挥抗糖尿病特性并导致代谢效率低下的原因,目前还缺乏一个可理解的解释。为了评估这个问题,我们系统地重新分析了已发表的来自 MPP 处理的神经元、二甲双胍处理的肝细胞和蛋氨酸限制的大鼠的转录组数据集。我们发现导致 NADPH 形成的途径与合成代谢脂肪酸生物合成一起被广泛诱导,后者在线粒体损伤的状态下显得高度矛盾。然而,同时诱导分解代谢脂肪酸氧化表明复合物 I 抑制产生了脂肪酸合成和降解的“无效”循环,该循环在体内解剖学上分布在脂肪组织和肝脏之间。辅因子平衡分析表明,这种循环确实在能量上是无效的(每个乙酰辅酶 A -3 ATP),尽管它不会是氧化还原无效的,因为它将 NADPH 转化为可呼吸的 FADH 2,而不产生任何 NADH。我们得出的结论是,抑制 NADH 脱氢酶会导致代谢从糖酵解和柠檬酸循环(均产生 NADH)转向戊糖磷酸途径,其产物 NADPH 通过脂肪酸循环以 1:1 的比例转化为 FADH 2 。肝脏和肠道复合物 I 抑制后的糖尿病抵抗表型归因于 FGF21 和 GDF15 依赖性脂肪饥饿信号,该信号将脂肪组织重塑为葡萄糖代谢器官。


























 京公网安备 11010802027423号
京公网安备 11010802027423号