Advanced Synthesis & Catalysis ( IF 5.4 ) Pub Date : 2023-09-22 , DOI: 10.1002/adsc.202300953 Carlos J. Moreno 1 , Samantha Gittings 2 , dieter schollmeyer 3 , Jesús Joglar 1 , Jordi Bujons 1 , Karel Hernández 4 , Pere Clapés 4
|
Chiral 2-hydroxy-4-arylbut-3-enoic acid derivatives are important precursors for the preparation of relevant biologically active compounds such as angiotensin converting enzyme (ACE) inhibitors (e. g. enalapril, lisinopril, cilapril or benazepril).1 Many efforts have been dedicated to develop strategies for the asymmetric synthesis of these type of molecules (Scheme 1).1

Asymmetric synthetic methodologies for the preparation of 2-hydroxy-4-arylbutanoic acid using 2-hydroxy- and 2-oxo-4-arylbut-3-enoic acid derivatives as intermediates. A) Enantio-complementary deracemization of (±)-2-hydroxy-4-phenylbut-3-enoic acid1a and B) asymmetric hydrogenation of 2-oxo-4-arylbut-3-enoic acid derivatives.1b-1d, 7
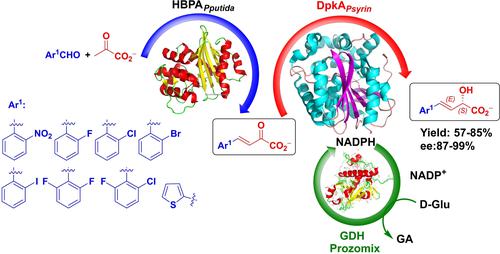
Asymmetric reduction on esters of 2-oxo-4-arylbut-3-enoic acids have a significant role in the synthesis of 2-hydroxy-4-arylbutyrates. However, these methods are laborious by the need to hydrolyze the ester and recrystallize the corresponding acids for improving their enantiomeric purity.1c, 2 Therefore, a more convenient alternative is to develop an efficient and asymmetric method for the reduction of 2-oxo-4-arylbut-3-enoic acids1c and then hydrogenation of the double bond by conventional methods. In this line of thinking, we focus our attention on Δ1-piperidine-2-carboxylate/Δ1-pyrroline-2-carboxylate reductase from Pseudomonas syringae pv. tomato DSM 50315 (DpkAPsyrin, EC 1.5.1.21), described as an imino reductase.3 In a previous work, we observed that DpkAPsyrin also exhibited promiscuous ketoreductase activity (Scheme 2A).4 Thus, we envision a straightforward enzymatic cascade process for the asymmetric synthesis of 2-hydroxy-4-arylbut-3-enoic acid derivatives. The strategy consisted of an aldol condensation reaction between pyruvate and aryl aldehydes catalyzed by trans-o-hydroxybenzylidene pyruvate hydratase-aldolase from Pseudomonas putida (HBPAPputida, EC 4.1.2.45)5 and an ensuing asymmetric reduction of the carbonyl group of the adduct catalyzed by DpkAPsyrin (Scheme 2B).

Enzymatic aldol-carbonyl reduction cascade reactions involving HBPAPputida and DpkAPsyrin catalysis. A) Aldol addition-reduction involving non-aromatic aldehydes described in a previous work;4 B) this work: aldol condensation-reduction involving aromatic aldehydes for the asymmetric synthesis of (S,E)-2-hydroxy-4-arylbut-3-enoic acids.
We previously reported that DpkAPsyrin was able to reduce carbonyl groups of different 2-oxoacid derivatives.4 In the previous work, products from the aldol addition of pyruvate to non-aromatic aldehydes obtained by HBPAPputida catalysis, were assayed with DpkAPsyrin. This was actually a promiscuous activity of HBPAPputida since in vivo this enzyme catalyzes the reversible aldol condensation between pyruvate and salicylaldehyde to yield trans-o-hydroxybenzylidenepyruvate.6 Thus, as a continuation of our synthetic study, we select aromatic aldehydes as electrophiles using pyruvate as donor component, furnishing arylbut-3-enoic acids, 3 a–p by HBPAPputida-catalyzed aldol condensation (Scheme 3).

Enzymatic cascade process for the synthesis of (S,E)-2-hydroxy-4-arylbut-3-enoic acid 4 comprising an aldol condensation with an ensuing C2 carbonyl reduction catalyzed by HBPAPputida and DpkAPsyrin, respectively. GDH, Glucose dehydrogenase was provided by Prozomix Ltd (PRO-GDH(001)). [a]Conversion of reduction reaction. No starting materials or aldol condensation products were detected by HPLC. [b]Isolated yields. [c]Enantiomeric excess were determined by HPLC on chiral stationary phases. [d]The product was obtained using a two-step synthesis: enzymatic reduction was conducted on the previously isolated (E)-2-oxo-4-phenylbut-3-enoic acid (3 a).
Compounds 3 a–p were submitted to reduction catalyzed by DpkAPsyrin. To establish reaction conditions, we begin with the biocatalytic reduction of previously isolated (E)-2-oxo-4-phenylbut-3-enoic acid (3 a) (see SI). The reaction proceeded in near quantitative conversion after 24 h (> 95%), furnishing 4 a with 62% isolated yield and 91% ee. However, when assaying the cascade process starting with pyruvate, benzaldehyde and both HBPAPputida and DpkAPsyrin, a 1:1 mixture of 3 a:4 a (determined by 1H NMR, see Figure S24) were obtained after purification. We reasoned that since pyruvate can be reduced to lactate by DpkAPsyrin (estimated kinetic parameters for pyruvate reduction Kmapp=101 mM and kcatapp=0.4 min−1, 1≤ [pyruvate] ≤60 mM, see Figure S10) this side reaction competes with 3 a for the NADPH regeneration lowering the yield. In addition, equilibrium limitations of the aldol condensation reaction can also affect the final yield of 4 a, when both HBPAPputida and DpkAPsyrin, are present in the reaction mixture. Thus, the successful cascade process will depend on both the efficiency of the aldol condensation (i. e., kinetics and reaction equilibrium) and the relative reduction rates of 2 and 3.
Gratifyingly, the cascade reactions using substrates 1 b–i rendered quantitative formation of the reduced product (4 b–i), with complete consumption of the starting material (1) and the intermediate precursors (3 b–i) (Scheme 3, panel A). Conversion was incomplete or even sluggish (35–61%) with aryl groups with meta and para substituents (e. g., 3 j–m) and were not scaled up (Scheme 3, panel B). On the other hand, DpkAPsyrin was inactive toward aryl groups with both metha and para substituents (e. g., 3 n), cinnamaldehyde (3 o) and 9H-fluorene-2-carbaldehyde (3 p), probably due to the steric limitations imposed by the active site cavity (vide infra).
After a work up and a purification by low-pressure reverse phase chromatography (Isolera Biotage® System, see SI), 4 b–i were obtained as carboxylic acids with isolated yields between 57–85% (Scheme 3).
Enantiomeric ratios of 2-hydroxy-4-arylbut-3-enoic acid (4) were determined by HPLC on a chiral stationary phase. The corresponding authentic racemic samples were prepared by Luche reduction8 from α,β-unsaturated 2-oxoacids (3 a–i) produced by HBPAPputida catalysis (see Figure S1 to S9). Excellent levels of enantioselectivity were achieved for 4 b, c, e–i (95–99% ee), and good for 4 d (87% ee). Single-crystal X-ray diffraction of 4 c–e indicated that DpkAPsyrin renders 2-hydroxyacids having an S configuration as the major stereoisomer (Figure 1). Consistent with our previous study,4 an S configuration can be safely assumed for the major enantiomers of 4 b, and 4 f–i (Scheme 3).

X-Ray structures of (E,S)-4 c, (E,S)-4 d, and (E,S)-4 e as ORTEP-type plots displaying one molecule with 50% probability ellipsoids. Compound (S,E)-4 c was observed with a disordered F-atom (ring-flipping). The data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data request/cif.
Molecular models of intermediates 3 bound into the active site of DpkAPsyrin (Figures 2 and S11) helped to understand our observations. As illustrated in Figure 2 for intermediate 3 b, these models show that the substrates adopt a disposition determined by the interactions of their carboxylate group with residues Arg58, Thr166, His192 and Gly193, and that between their 2-oxo group and the protonated imidazole of His54, which acts as a general acid catalyst.4

Molecular model of intermediate 3 b bound into the active site of DpkAPsyrin. This model was built starting from PDB structure 2CWH through optimization by QM/MM methods, with QSite4, 9 at the DFT B3LYP/6–31G** level of theory, as previously described.4 The substrate, NADPH and close protein residues are shown with yellow, green and gray C-atoms; H-bonds, salt bridges and π-stacking interactions are shown with yellow, cyan and magenta dashed lines.
This substrate binding mode fixes the pre-reactive conformation and forces the exposure of the re-face of the 2-oxo group to the reduced NADPH. The aromatic rings present in substrates 3 are stabilized by π-stacking interactions with the imidazole group of His192, and their substituents in ortho-position (3 b–f) display a preferred orientation towards Arg58 since the opposite would place them too close to residues 150–151. Only the ortho-fluoride seems to be small enough to fit in an orientation towards these two residues, as demonstrated by the results obtained for compounds 3 g and 3 h. Substrates with meta or para-substituents (3 j–n), or with larger aromatic substituents (3 o–p), are not well accepted because of the steric restrictions imposed by the proximity of residues 150–153 and 263.
In summary, we expanded the synthetic application of the promiscuous ketoreductase activity of DpkAPsyrin for the asymmetric reduction of 2-oxo-4-arylbut-3-enoic acid derivatives. The cascade enzymatic system using HBPAPputida as catalyst for the construction of carbon scaffold and DpkAPsyrin for carbonyl reduction provide homochiral (S,E)-2-hydroxy-4-arylbut-3-enoic acid derivatives with diverse aromatic moieties from achiral starting material in 57%-85% isolated yields and ee 87%-99%. DpkAPsyrin gave quantitative conversions after 24 h of incubation tolerating substrates with substitutions at ortho positions of the aromatic ring. Incomplete conversions were observed with aryl groups bearing substituents with meta and para substituents, whereas it was completely inactive towards substrates bearing aryl groups with both ortho and para substituents, 2-oxo-dienoic acids and fused aromatic rings.
Experimental Section
Materials
Benzaldehyde (1 a), 2-nitrobenzaldehyde (1 b), 2-fluorobenzaldehyde (1 c), 2-chlorobenzaldehyde (1 d), 2-iodobenzaldehyde (1 f), 2,6-fluorbenzaldehyde (1 g), 2-chloro-6-fluorobenzaldehyde (1 h), thiophene-3-carboxaldehyde (1 i), 3-nitrobenzaldehyde (1 j), 4-nitrobenzaldehyde (1 k), 3-chlorobenzaldehyde (1 l), 4-chlorobenzaldehyde (1 m), 2-Hydroxy-3-methoxybenzaldehyde (1 n), trans-cinnamaldehyde (1 o), fluorene-2-carboxaldehyde (1 p), indole-3-carboxaldehyde (1 q) and sodium pyruvate (2 a) were purchased from Sigma-Aldrich. 2-bromobenzaldehyde (1 e), sodium borohydride were purchased from TCI chemical. NADPH was purchased from CARL ROTH. Glucose dehydrogenase (GDH) as a cell free extract powder and NADP+ were provided by Prozomix Ltd (PRO-GDH(001)). Water for analytical HPLC was obtained from an Arium pro ultrapure water purification system (Sartorius Stedim Biotech) and the rest of solvents used in this work were of analytical grade or HPLC grade.
Methods
Enzyme Production
General procedure for, HBPAPputida, GDH and DpkAPsyrin expression, purification and activity determination were performed as describe in previous works (Table S1).10
HPLC Analysis
HPLC analysis was performed on a RP-HPLC XBridge® C18, 5 μm, 4.6×250 mm column (Waters). The solvent system used was: solvent (A): 0.1% (v/v) trifluoroacetic acid (TFA) in H2O and solvent (B): 0.095% (v/v) TFA in CH3CN/H2O 4:1, flow rate 1 mL min−1, detection at 215 nm and column temperature at 30 °C. Substrates and products were quantified from the peak areas using an external standard methodology. Reaction monitoring for benzaldehyde (1 a), aldol adduct (3) and reduced product (4) was carried out as follows: samples were withdrawn from the reaction mixture (25 μL) and diluted with methanol (500 μL). After centrifugation, samples were analyzed by HPLC. Elution conditions: gradient from 10 to 100% B over 30 min.
Enantiomeric excesses were analyzed by HPLC on a CHIRALPAK® ID, column (46×250 mm, 5 μm). The solvent system used was: solvent (A): 0.1%TFA in hexane and solvent (B):0.1%TFA in isopropanol, detection by diode array detection (215–350 nm) and column temperature at 30 °C.
NMR Analysis
Routine 1H (400 MHz) and 13C (101 MHz) NMR spectra of compounds were recorded with a Varian Mercury-400 spectrometer. Full characterization of the described compounds was performed using typical gradient-enhanced 2D experiments: COSY, HSQC, and NOESY recorded under routine conditions.
Biocatalytic Aldol Condensation Between 2 and Aldehydes (1 a–p), Catalyzed by HBPAPputida and Variant
Analytical scale: The reaction (total volume 500 μL) was carried out in 1.5 mL Eppendorf tubes. To a solution of the aldehydes (1 a–p) dissolved in DMF (20% v/v in the reaction), a solution of sodium pyruvate (2) (25 μL of a 2.0 M aqueous stock solution, pH 6.5–7.0, final concentration 100 mM) was added in the reaction. The reaction was initiated with the addition of HBPAPputida (125 μL of a 0.029 U mL−1 stock solution, 4 mg mL−1 in 50 mM TEA buffer, 50 mM NaCl, 0.5 mM EDTA and 50% (v/v) glycerol, 0.007 U mL−1, 1 mg mL−1 protein mL−1 final concentration in the reaction) (Table S2). The reaction mixture was placed on a vortex mixer (1000 rpm) at 25 °C for 24 h. Samples were withdrawn immediately after enzyme addition (0 h) and after 24 h and analyzed by HPLC as described above.
Biocatalytic Reduction of arylbut-3-enoic Acids (3 a–p) Catalyzed by DpkAPsyrin. Analytical Scale
Reactions were carried out at analytic level as follows: The reactions (500 μL total volume) were conducted in Eppendorf tubes (1.5 mL) and placed in a vortex mixer (1000 rpm) at 25 °C. To a solution of the aldehydes (1 a–p) dissolved in DMF (20% v/v in the reaction), a solution of sodium pyruvate (2) (25 μL of a 2.0 M aqueous stock solution, pH 6.5–7.0, final concentration 100 mM), DpkAPsyrin (125 μL of a stock solution of 2.6 10−2 U mL−1, 4 mg mL−1 in 20 mM TEA buffer pH 7.0, 100 mM NaCl, and 50% (v/v) of glycerol, 6.5 10−3 U mL−1 final concentration in the reaction), GDH (83 μL of a stock solution 20.8 U mL−1, 5.2 mg mL−1 in 10 mM HEPES buffer pH 6.5, 50 mM NaCl, and 50% (v/v) of glycerol, 3.5 U mL−1 final concentration in the reaction), a solution of NADP+ and glucose (42 μL, NADP+ stock solution 60 mM, 5 mM final concentration in the reaction, glucose stock solution 2.4 M, 200 mM in reaction, dissolved in 1 M sodium phosphate buffer pH 8.0, 84 mM final concentration in the reaction). The reaction was initiated with the addition of HBPAPputida (125 μL of a 0.029 U mL−1 stock solution, 4 mg mL−1 in 50 mM TEA buffer, 50 mM NaCl, 0.5 mM EDTA and 50% (v/v) glycerol, 0.007 U mL−1, 1 mg mL−1 protein mL−1 final concentration in the reaction) (Table S3). Samples were withdrawn immediately after the addition of aldol substrate (0 h) and after 24 h and analyzed by HPLC as described above.
Synthesis of arylbut-3-enoic Acids (3)(E)-2-oxo-4-phenylbut-3-enoic Acid (3 a)

Synthesis of (E)-2-oxo-4-phenylbut-3-enoic acid (3 a). Typical procedure: The reaction (0.5 mmol scale, 20 mL total volume) was conducted in an Erlenmeyer at 25 °C and an orbital shaker (250 rpm). To a solution of the aldehyde (1 a, 110.0 mg, 1.04 mmol, 50 mM in reaction) dissolved in DMF (20% v/v in reaction), a solution of sodium pyruvate (2) (1.5 mL of a 2.0 M concentrated solution, pH 6.5–7.0, 3.12 mmol, 150 mM in reaction, 3 eq) was added. The reaction was initiated with the addition of enzyme (HBPAPputida, 5 mL of a 0.029 U mL−1 stock solution, 4 mg mL−1 in 50 mM TEA buffer, 50 mM NaCl, 0.5 mM EDTA and 50% (v/v) glycerol, 0.007 U mL−1, 1 mg mL−1 protein mL−1 final concentration in reaction). The mixture was placed on an orbital shaker (250 rpm) at 25 °C for 24 h. The reaction was monitored by HPLC as described above. After completion of the reaction the enzyme component was precipitated with MeOH (10 volumes) filtered over Celite® the filtrate was washed with 5% NaHCO3, (3×100 mL), the organic solvent was evaporated and the aqueous phase was extracted with AcOEt (3×100 mL). The pH of the aqueous phase was adjusted to pH 2.0 with 3 M HCl. The compound was extracted with AcOEt (3×100 mL). This organic phase was washed with pure H2O (3×100 mL), then with saturated NaCl solution (3×100 mL), dried over anhydrous MgSO4 and concentrated under vacuum. The solid was adsorbed on a KP−C18-HS SNAP Cartridge and purified by Biotage Isolera with gradient (solvent system used was A: H2O+0.1% HCO2H and B: CH3CN+0.1% HCO2H), from 0 to 100% B, 20 VC and 100% B 10 VC. After purification, the pure fractions were collected and the organic solvent was removed by vacuum, the compound was extracted from the acidic aqueous phase with AcOEt (3×50 mL), washed with pure H2O (3×50 mL), then with saturated NaCl solution (3×50 mL), dried over anhydrous MgSO4 and concentrated under vacuum, to afford compound 3 a as a yellow solid (161.2 mg, 81%). 1H NMR (400 MHz, DMSO) δ 7.88 – 7.78 (m, 2H), 7.76 (d, J=16.3 Hz, 1H), 7.56 – 7.40 (m, 3H), 7.30 (d, J=16.3 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 186.6, 164.9, 148.0, 134.3, 132.0, 129.6, 122.0. The spectral properties of this product agreed with those reported in the literature.5, 6
(E)-4-(2-nitrophenyl)-2-oxobut-3-enoic Acid (3 b)

The title compound was prepared as described for 3 a. Starting from 1 b ([1 b]=150.0 mg, 50 mM and [2]=327.7 mg, 150 mM in the reaction), 3 b was obtained as an orange solid (183.9 mg, 84%). 1H NMR (400 MHz, DMSO) δ 8.12 (dd, J=8.1, 1.3 Hz, 1H), 8.04 (d, J=16.2 Hz, 1H), 8.00 (d, J=8.0 Hz, 1H), 7.82 (td, J=7.7, 7.3, 0.7 Hz, 1H), 7.73 (ddd, J=8.8, 7.5, 1.4 Hz, 1H), 7.27 (d, J=16.1 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 185.7, 163.9, 148.6, 142.1, 134.1, 131.7, 129.3, 125.2. The spectral properties of this product matched those reported in the literature.5
(E)-4-(2-fluorophenyl)-2-oxobut-3-enoic Acid (3 c)

The title compound was prepared as described for 3 a. Starting from 1 c ([1 c]=110.0 mg, 50 mM and [2]=292.6 mg, 150 mM in the reaction), 3 c was obtained as a yellow solid (87.5 mg, 51%).1H NMR (400 MHz, CDCl3) δ 8.30 (d, J=16.3 Hz, 1H), 7.77 – 7.71 (m, 2H), 7.51 (dddd, J=8.9, 7.3, 5.3, 1.7 Hz, 1H), 7.31 – 7.24 (m, 1H), 7.19 (ddd, J=10.6, 8.3, 1.2 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 182.5, 162.4 (d, J=257.3 Hz), 160.1, 143.7 (d, J=3.5 Hz), 134.2 (d, J=9.0 Hz), 129.9 (d, J=2.2 Hz), 125.0 (d, J=3.7 Hz), 122.2 (d, J=11.2 Hz), 119.6 (d, J=6.5 Hz), 116.7 (d, J=21.8 Hz).5
(E)-4-(2-chlorophenyl)-2-oxobut-3-enoic Acid (3 d)

The title compound was prepared as described for 3 a. Starting from 1 d ([1 d]=150.0 mg, 50 mM and [2]=352.3 mg, 150 mM in the reaction), 3 d was obtained as a yellow solid (179.1 mg, 80%).1H NMR (400 MHz, DMSO) δ 8.03 (dd, J=7.1, 2.4 Hz, 1H), 8.02 (d, J=16.8 Hz, 1H), 7.59 (dd, J=8.0, 1.4 Hz, 1H), 7.52 (td, J=8.0, 7.6, 1.7 Hz, 1H), 7.44 (td, J=2x7.8, 0.9 Hz, 1H), 7.37 (d, J=16.2 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 185.7, 164.0, 141.6, 134.6, 132.9, 131.4, 130.2, 128.6, 128.0, 124.2. The spectral properties of this product matched those reported in the literature.5
(E)-4-(2-bromophenyl)-2-oxobut-3-enoic Acid (3 e)

The title compound was prepared as described for 3 a. Starting from 1 e ([1 e]=150.0 mg, 50 mM and [2]=267.7 mg, 150 mM in the reaction), 3 d was obtained as a yellow solid (183.8 mg, 89%). 1H NMR (400 MHz, DMSO) δ 8.02 (d, J=1.9 Hz, 1H), 7.98 (d, J=16.0 Hz, 1H), 7.76 (dd, J=7.9, 1.4 Hz, 1H), 7.48 (td, J=2x7.6, 1.4 Hz, 1H), 7.42 (td, J=7.6, 7.4, 1.8 Hz, 1H), 7.33 (d, J=16.2 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 186.2, 164.5, 144.8, 134.0, 133.6, 133.5, 129.2, 129.0, 126.1, 124.8.
(E)-4-(2-iodophenyl)-2-oxobut-3-enoic Acid (3 f)

The title compound was prepared as described for 3 a. Starting from 1 f ([1 f]=160.0 mg, 50 mM and [2]=227.7 mg, 150 mM in the reaction), 3 f was obtained as a yellow solid (160.3 mg, 77%). 1H NMR (400 MHz, DMSO) δ 8.00 (dd, J=7.9, 1.2 Hz, 1H), 7.94 (dd, J=7.9, 1.6 Hz, 1H), 7.87 (d, J=16.1 Hz, 1H), 7.48 (td, J=7.7, 7.6, 1.1 Hz, 1H), 7.24 (d, J=16.1 Hz, 1H), 7.22 (td, J=7.6, 7.5, 1.6 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 185.8, 164.2, 149.6, 140.0, 136.2, 132.8, 129.0, 128.2, 124.2, 103.4. The spectral properties of this product matched those reported in the literature.5
(E)-4-(2,6-difluorophenyl)-2-oxobut-3-enoic Acid (3 g)

The title compound was prepared as described for 3 a. Starting from 1 g ([1 g]=150.0 mg, 50 mM and [2]=348.5 mg, 150 mM in the reaction), 3 g was obtained as a yellow solid (174.6 mg, 78%). 1H NMR (400 MHz, DMSO) δ 7.68 (d, J=16.7 Hz, 1H), 7.61 (t, J=2x8.5 Hz, 1H), 7.46 (d, J=16.6 Hz, 1H), 7.28 (t, J=2x8.9 Hz, 2H). 13C NMR (101 MHz, DMSO) δ 185.0, 163.4, 161.2(dd, J=254.7, 6.6 Hz), 133.6 (t, J=2x11.3, Hz), 131.7, 126.5 (t, J=2x8.4, Hz), 112.7, 112.4, 111.33 (t, J=2x14.8 Hz).
(E)-4-(2-chloro-6-fluorophenyl)-2-oxobut-3-enoic Acid (3 h)

The title compound was prepared as described for 3 a. Starting from 1 h ([1 h]=150.0 mg, 50 mM and [2]=312.3 mg, 150 mM in the reaction), 3 h was obtained as a yellow solid (144.2 mg, 67%). 1H NMR (400 MHz, DMSO) δ 7.82 (d, J=16.6 Hz, 1H), 7.60 – 7.49 (m, 2H), 7.47 (d, J=6.3 Hz, 1H), 7.44 (d, J=16.5 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 185.1, 163.4, 161.6 (d, J=255.9 Hz), 135.6 (d, J=2.0 Hz), 135.40 (d, J=5.1 Hz), 133.0 (d, J=10.6 Hz), 127.6 (d, J=14.2 Hz), 126.6 (d, J=3.2 Hz), 120.5 (d, J=14.0 Hz), 115.8 (d, J=22.9 Hz).
(E)-2-oxo-4-(thiophen-2-yl)but-3-enoic Acid (3 i)

The title compound was prepared as described for 3 a. Starting from 1 i ([1 i]=120.0 mg, 50 mM and [2]=353.3 mg, 150 mM in the reaction), 3 i was obtained as a yellow solid (184.8 mg, 95%). 1H NMR (400 MHz, DMSO) δ 7.94 (d, J=16.0 Hz, 1H), 7.87 (d, J=5.0 Hz, 1H), 7.72 (d, J=0.6 Hz, 1H), 7.22 (dd, J=5.0, 3.7 Hz, 1H), 6.97 (d, J=15.9 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 184.9, 164.2, 139.9, 139.1, 134.6, 132.1, 129.1, 119.6.
(E)-4-(3-chlorophenyl)-2-oxobut-3-enoic Acid (3 l)

The title compound was prepared as described for 3 a. Starting from 1 l ([1 l]=150.0 mg, 50 mM and [2]=352.3 mg, 150 mM in the reaction), 3 l was obtained as a yellow solid (94.4 mg, 42%). 1H NMR (400 MHz, DMSO) δ 7.94 (t, J=2x1.9 Hz, 1H), 7.79 (dt, J=7.6, 2x1.4 Hz, 1H), 7.72 (d, J=16.3 Hz, 1H), 7.59 – 7.49 (m, 1H), 7.48 (t, J=2x7.8 Hz, 1H), 7.36 (d, J=16.3 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 186.4, 164.3, 145.5, 136.1, 133.9, 131.0, 130.9, 128.6, 127.6, 123.1.5
(E)-4-(4-chlorophenyl)-2-oxobut-3-enoic Acid (3 m)

The title compound was prepared as described for 3 a. Starting from 1 m ([1 m]=150.0 mg, 50 mM and [2]=352.3 mg, 150 mM in the reaction), 3 l was obtained as a yellow solid (163.3 mg, 73%). 1H NMR (400 MHz, DMSO) δ 7.85 (d, J=8.6 Hz, 2H), 7.74 (d, J=16.3 Hz, 1H), 7.53 (d, J=8.5 Hz, 2H), 7.31 (d, J=16.3 Hz, 1H).13C NMR (101 MHz, DMSO) δ 186.7, 164.9, 146.3, 136.5, 133.3, 131.6, 131.3, 129.6, 129.2, 122.8. The spectral properties of this product matched those reported in the literature.5
(E)-4-(1H-indol-3-yl)-2-oxobut-3-enoic Acid (3 q)

The title compound was prepared as described for 3 a. Starting from 1 q ([1 q]=130.0 mg, 50 mM and [2]=295.7 mg, 150 mM in the reaction), 3 q was obtained as a yellow solid (129.9 mg, 67%). 1H NMR (400 MHz, DMSO) δ 8.17 (d, J=3.0 Hz, 1H), 8.00 (d, J=16.0 Hz, 1H), 7.90 (dd, J=6.5, 2.4 Hz, 1H), 7.51 (dd, J=7.5, 1.7 Hz, 1H), 7.25 (tt, J=2x7.2, 2x5.5 Hz, 2H), 7.10 (d, J=16.0 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 166.0, 143.9, 138.2, 135.4, 125.4, 123.6, 122.2, 120.6, 115.3, 113.3.
Synthesis of (S,E)-2-hydroxy-4-arylbut-3-enoic Acid Derivatives (S-4) and their Racemates (rac-4)
(S,E)-2-hydroxy-4-(2-nitrophenyl)but-3-enoic Acid (S-4 b)

Typical procedure: the reaction (0.5 mmol scale, 20 mL total volume) was conducted in a round-bottom flask (100 mL) at 25 °C and magnetically stirred with a bar at 250 rpm. To a solution of the aldehyde (1 b, 150.0 mg, 1 mmol, 1 eq, 50 mM in the reaction) dissolved in DMF (4.0 mL, 20% (v/v) in the reaction), a solution of sodium pyruvate (2) (500 μL of a 2.0 M aqueous stock solution, pH 6.5–7.0, final concentration 50 mM), HBPAPputida (5.0 mL of a 0.029 U mL−1 stock solution, 4 mg mL−1 in 50 mM TEA buffer, 50 mM NaCl, 0.5 mM EDTA and 50% (v/v) glycerol, 0.007 U mL−1, 1 mg mL−1 protein mL−1 final concentration in the reaction), glucose (720.6 mg, 2 mmol, 4 eq, 0.2 M final concentration in the reaction), GDH (3.3 mL of a stock solution 20.8 U mL−1, 5.2 mg mL−1 in 10 mM HEPES buffer pH 6.5, 50 mM NaCl, and 50% (v/v) of glycerol, 3.4 U mL−1 final concentration in the reaction) and DpkAPsyrin (5.0 mL of a stock solution 2.6 10−2 U mL−1, 4 mg mL−1 in 20 mM TEA buffer pH 7.0, 100 mM NaCl, and 50% (v/v) of glycerol, 6.5 10−3 U mL−1 final concentration in the reaction) were adding. The reaction was started by adding a solution of NADP+ (2.2 mL of stock solution 16 mM in 0.8 M sodium phosphate buffer pH 8.0, 5 mM final concentration in the reaction). Samples were withdrawn immediately after the addition of aldol substrate (0 h) and after 24 h and analyzed by HPLC as described above. The purification was done following the protocol described for 3 a. S-4 b was obtained as a white solid (152.2 mg, 69%), [α]20D=+35.9 (c=1, in MeOH).1H NMR (400 MHz, DMSO) δ 7.94 (dd, J=8.1, 1.3 Hz, 1H), 7.80 (dd, J=7.9, 1.4 Hz, 1H), 7.69 (td, J=7.7, 7.6, 1.4 Hz, 1H), 7.53 (td, J=7.8, 7.4, 1.4 Hz, 1H), 6.99 (dd, J=15.8, 2.0 Hz, 1H), 6.51 (dd, J=15.7, 4.9 Hz, 1H), 4.77 (dd, J=4.9, 2.0 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 173.2, 147.9, 133.5, 133.3, 130.6, 128.7, 128.2, 124.1, 123.9, 70.6. ESI-TOF m/z: Calcd for [M+Na+] C10H9NO5Na+: 246.0373, found [M+Na+]: 246.0365.
(E)-2-hydroxy-4-(2-nitrophenyl)but-3-enoic Acid (rac-4 b)

The aldol precursor 3 b was prepared and purified as described for 3 a, starting from 1 b (150.0 mg, 1 eq, 0.1 M concentration in the reaction) and 2 (109.2 mg, 1 eq, 0.1 M concentration in the reaction). Chemical reduction was carried out as follows: Typical procedure: A dried three-necked round bottomed flask was charged with anhydrous MeOH (25 mL) under N2 atmosphere. Then, CeCl3*7H2O (6.6 g, 17.2 mmol, 39.2 eq) was dissolved, and added a portion of 3 b (100.0 mg, 0.45 mmol, 1.0 eq) after 30 minutes on magnetic stirring (300 rpm) at rt. The chemical reduction was initiated by adding NaBH4 (17.1 mg, 0.45 mmol, 1.0 eq), the mixture was stirred at 25 °C for 10 min. Formation of rac-4 b was estimated by measuring the aldol adduct 3 b consumed by HPLC. When the conversion was maximum, a solution 5% NaHCO3 (50 mL) was add to stop the reaction. After that, the solvent was evaporated under vacuum. Aqueous phase was adjusted to pH 2.0 with a 3 M HCl solution and extracted with AcOEt (3×50 mL). The organic phases were combined, washed with water (3×100 mL), brine (3×100 mL) and dried over anhydrous magnesium sulfate. The solvent removed under vacuum affording (E)-2-hydroxy-4-(2-nitrophenyl)but-3-enoic acid (rac-4 b) as brown solid (85.4 mg, 85%). NMR spectra were indistinguishable from that of S-4 b.
(S,E)-4-(2-fluorophenyl)-2-hydroxybut-3-enoic Acid (S-4 c)

The title compound was prepared as described for S-4 b, starting from 1 c (110.0 mg) S-4 c was obtained as a white solid (138.3 mg, 72%). [α]20D=+67.2 (c=1, in MeOH). 1H NMR (400 MHz, DMSO) δ 7.60 (t, J=7.3, 7.3 Hz, 1H), 7.31 (dd, J=9.9, 3.9 Hz, 1H), 7.19 (q, J=7.8, 7.8, 7.2 Hz, 1H), 6.82 (d, J=16.1 Hz, 1H), 6.48 (dd, J=16.1, 5.2 Hz, 1H), 4.72 (dd, J=5.1, 1.4 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 173.5, 159.5 (d, J=247.2 Hz), 130.9 (d, J=4.7 Hz), 129.4 (d, J=8.5 Hz), 127.7 (d, J=3.6 Hz), 124.7 (d, J=3.3 Hz), 123.7 (d, J=11.9 Hz), 122.0 (d, J=3.7 Hz), 115.7 (d, J=21.9 Hz), 70.8. ESI-TOF m/z: Calcd for [M+Na+] C10H9FO3Na+: 219.0428, found [M+Na+]: 219.0425.
(E)-4-(2-fluorophenyl)-2-hydroxybut-3-enoic Acid (rac-4 c)

Synthesis of title compound was performed as described for rac-4 b starting from 3 c (70.0 mg). rac-4 c was obtained as a white solid (59.2 mg, 84%). NMR spectra were indistinguishable from that of S-4 c.
(S,E)-4-(2-chlorophenyl)-2-hydroxybut-3-enoic Acid (S-4 d)

The title compound was prepared as described for S-4 b, starting from 1 d (130.0 mg). S-4 d was obtained as a white solid (140.1 mg, 71%). [α]20D=+39.6 (c=1, in MeOH). 1H NMR (400 MHz, DMSO) δ 7.68 (dd, J=7.5, 2.0 Hz, 1H), 7.45 (dd, J=7.5, 1.8 Hz, 1H), 7.30 (pd, J=4x7.3, 1.8 Hz, 2H), 7.03 (dd, J=15.9, 1.9 Hz, 1H), 6.45 (dd, J=15.8, 5.1 Hz, 1H), 4.75 (dd, J=5.1, 2.0 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 173.4, 134.0, 131.9, 131.3, 129.6, 129.2, 127.5, 127.1, 125.3, 70.7. ESI-TOF m/z: Calcd for [M+Na+] C10H9ClO3Na+: 235.0132, found [M+Na+]: 235.0122.
(E)-4-(2-chlorophenyl)-2-hydroxybut-3-enoic Acid (rac-4 d)

Synthesis of title compound was performed as described for rac-4 b starting from 3 d (100.0 mg), rac-4 d was obtained as a white solid (95.2 mg, 95%). NMR spectra were indistinguishable from that of S-4 d.
(S,E)-4-(2-bromophenyl)-2-hydroxybut-3-enoic Acid (S-4 e)

The title compound was prepared as described for S-4 b, starting from 1 e (150.0 mg). S-4 e was obtained as a white solid (163.2 mg, 78%). [α]20D=+30.1 (c=1, in MeOH).1H NMR (400 MHz, DMSO) δ 7.66 (d, J=6.2 Hz, 1H), 7.62 (d, J=8.0 Hz, 1H), 7.36 (t, J=2x6.9, Hz, 1H), 7.20 (td, J=7.7, 7.6, 1.7 Hz, 1H), 6.99 (dd, J=15.8, 1.9 Hz, 1H), 6.41 (dd, J=15.8, 5.1 Hz, 1H), 4.75 (dd, J=5.2, 2.0 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 173.4, 135.7, 132.9, 131.4, 129.5, 128.1, 128.0, 127.2, 122.9, 70.6. ESI-TOF m/z: Calcd for [M+Na+] C10H9BrO3Na+: 278.9633, found [M+Na+]: 278.9639.
(E)-4-(2-bromophenyl)-2-hydroxybut-3-enoic Acid (rac-4 e)

Synthesis of title compound was performed as described for rac-4 b starting from 3 e (100.0 mg), rac-4 e was obtained as a white solid (90.4 mg, 90%). NMR spectra were indistinguishable from that of S-4 e.
(S,E)-2-hydroxy-4-(2-iodophenyl)but-3-enoic Acid (S-4 f)

The title compound was prepared as described for S-4 b, starting from 1 f (160.0 mg). S-4 f was obtained as a white solid (158.3 mg, 75%). [α]20D=+30.6 (c=1, in MeOH). 1H NMR (400 MHz, DMSO) δ 7.87 (dd, J=7.9, 1.3 Hz, 1H), 7.59 (dd, J=7.9, 1.7 Hz, 1H), 7.37 (td, J=2x7.6, 1.3 Hz, 1H), 7.02 (td, J=2x7.6, 1.6 Hz, 1H), 6.86 (dd, J=15.7, 1.9 Hz, 1H), 6.33 (dd, J=15.7, 5.1 Hz, 1H), 4.74 (dd, J=5.1, 2.0 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 173.4, 139.4, 138.9, 133.0, 131.2, 129.5, 128.7, 126.6, 100.2, 70.5. ESI-TOF m/z: Calcd for [M+Na+] C10H9IO3Na+: 326.9494, found [M+Na+]: 326.9485.
(E)-2-hydroxy-4-(2-iodophenyl)but-3-enoic Acid (rac-4 f)

Synthesis of title compound was performed as described for rac-4 b starting from 3 f (100.0 mg), rac-4 f was obtained as a white solid (98.1 mg, 98%). NMR spectra were indistinguishable from that of S-4 f.
(S,E)-4-(2,6-difluorophenyl)-2-hydroxybut-3-enoic Acid (S-4 g)

The title compound was prepared as described for S-4 b, starting from 1 g (130.0 mg). S-4 g was obtained as a white solid (138.3 mg, 70%). [α]20D=+65.3 (c=1, in MeOH). 1H NMR (400 MHz, DMSO) δ 7.36 (tt, J=8.5, 8.5, 6.5, 6.5 Hz, 1H), 7.13 (t, J=8.6, 8.6 Hz, 2H), 6.73 (dd, J=16.3, 1.8 Hz, 1H), 6.61 (dd, J=16.4, 4.7 Hz, 1H), 4.77 (dd, J=4.7, 1.8 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 173.3, 160.6 (dd, J=249.5, 7.8 Hz), 135.1 (t, J=7.6, 7.6 Hz), 129.3 (t, J=2x10.9, Hz), 115.8, 113.1, 112.1 (d, J=6.3 Hz), 111.9 (d, J=6.2 Hz), 70.9. ESI-TOF m/z: Calcd for [M+Na+] C10H9F2O3Na+: 237.0339, found [M+Na+]: 237.0341.
(E)-4-(2,6-difluorophenyl)-2-hydroxybut-3-enoic Acid (rac-4 g)

Synthesis of title compound was performed as described for rac-4 b starting from 3 g (100.0 mg), rac-4 g was obtained as a yellow solid (90.1 mg, 90%). NMR spectra were indistinguishable from that of S-4 g.
(S,E)-4-(2-chloro-6-fluorophenyl)-2-hydroxybut-3-enoic Acid (S-4 h)

The title compound was prepared as described for S-4 b, starting from 1 g (130.0 mg). S-4 h was obtained as a white solid (160.1 mg, 85%). [α]20D=+46.6 (c=1, in MeOH). 1H NMR (400 MHz, DMSO) δ 7.36 (td, J=7.9, 7.8, 2.1 Hz, 2H), 7.33 – 7.22 (m, 1H), 6.83 (dd, J=16.3, 2.0 Hz, 1H), 6.57 (ddd, J=16.3, 4.7, 1.1 Hz, 1H), 4.78 (dd, J=4.8, 2.0 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 173.3, 160.5 (d, J=250.7 Hz), 136.0 (d, J=11.8 Hz), 133.22 (d, J=5.7 Hz), 129.4 (d, J=10.4 Hz), 125.9 (d, J=3.4 Hz), 122.6 (d, J=14.7 Hz), 119.7, 115.2 (d, J=23.4 Hz), 70.8. ESI-TOF m/z: Calcd for [M+Na+] C10H9ClFO3Na+: 230.0146, found [M+Na+]: 230.0141.
(E)-4-(2-chloro-6-fluorophenyl)-2-hydroxybut-3-enoic Acid (rac-4 h)

Synthesis of title compound was performed as described for rac-4 b starting from 3 h (100.0 mg), rac-4 h was obtained as a white solid (90.2 mg, 90%). NMR spectra were indistinguishable from that of S-4 h.
(S,E)-2-hydroxy-4-(thiophen-2-yl)but-3-enoic Acid (S-4 i)

The title compound was prepared as described for S-4 b, starting from 1 f (100.0 mg). S-4 i was obtained as a white solid (94.4 mg, 57%). [α]20D=+107.5 (c=0.5, in MeOH). 1H NMR (400 MHz, DMSO) δ 7.42 (d, J=5.0 Hz, 1H), 7.11 (d, J=3.5 Hz, 1H), 7.01 (dd, J=5.1, 3.5 Hz, 1H), 6.85 (dd, J=15.7, 1.8 Hz, 1H), 6.08 (dd, J=15.7, 5.6 Hz, 1H), 4.64 (dd, J=5.6, 1.8 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 173.6, 141.1, 127.8, 127.2, 126.5, 125.2, 123.7, 70.5. ESI-TOF m/z: Calcd for [M+Na+] C8H8NaO3S: 207.0092, found [M+Na+]: 207.0083.
(E)-2-hydroxy-4-(thiophen-2-yl)but-3-enoic Acid (rac-4 i)

Synthesis of title compound was performed as described for rac-4 b starting from 3 i (45.0 mg), rac-4 i was obtained as a white solid (43.0 mg, 96%). NMR spectra were indistinguishable from that of S-4 i.
Synthesis of S-4 a in Two-Step

Aldol addition (1st): Synthesis of 3 a. The reaction (1 mmol scale, 14 mL total volume) was conducted in an erlenmeyer at 25 °C and an orbital shaker (250 rpm). To a solution of the aldehyde (1 a, 150.0 mg, 1.4 mmol, 100 mM in reaction) dissolved in DMF (20% v/v in reaction), a solution of sodium pyruvate (2) (700 μL of a 2.0 M concentrated solution, pH 6.5–7.0, 3.12 mmol, 100 mM in reaction, 1eq) was added. The reaction was initiated with the addition of enzyme (HBPAPputida, 3.5 mL of a 0.029 U mL−1 stock solution, 4 mg mL−1 in 50 mM TEA buffer, 50 mM NaCl, 0.5 mM EDTA and 50% (v/v) glycerol, 0.007 U mL−1, 1 mg mL−1 protein mL−1 final concentration in reaction). The mixture was placed on an orbital shaker (250 rpm) at 25 °C for 24 h. The reaction was monitored by HPLC as described above. Aldol reduction (2nd). After 24 h, the reduction reaction (28 mL final volume) was carried out adding glucose (1 g, 4.0 mmol, 4 eq, 0.2 M final concentration in the reaction,), GDH (4.6 mL of a stock solution 20.8 U mL−1, 5.2 mg mL−1 in 10 mM HEPES buffer pH 6.5, 50 mM NaCl, and 50% (v/v) of glycerol, 3.4 U mL−1 final concentration in the reaction), and DpkAPsyrin (7.0 mL of a stock solution 2.6 10−2 U mL−1, 4 mg mL−1 in 20 mM TEA buffer pH 7.0, 100 mM NaCl, and 50% (v/v) of glycerol, 6.5 10−3 U mL−1 final concentration in the reaction) were adding. The reaction was started by adding a solution of NADP+ (2.4 mL of stock solution 58.3 mM in 1 M sodium phosphate buffer pH 8.0, 5 mM final concentration in the reaction) and monitored by HPLC as described above. The purification was done following the protocol described for 3 a. the compound S-4 a was obtained as a white solid (176.2 mg, 62%). 1H NMR (400 MHz, DMSO) δ 7.43 (d, J=6.9 Hz, 1H), 7.33 (t, J=2x7.6 Hz, 2H), 7.25 (t, J=2x7.3 Hz, 1H), 6.69 (dd, J=16.0, 1.8 Hz, 1H), 6.35 (dd, J=15.9, 5.6 Hz, 1H), 4.68 (dd, J=5.6, 1.8 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 173.8, 136.2, 130.2, 128.7, 127.9, 127.7, 126.4, 70.9. ESI-TOF m/z: Calcd for [M+Na+] C10H10O3Na+: 201.0528, found [M+Na+]: 201.0534.
(E)-2-hydroxy-4-phenylbut-3-enoic Acid (rac-4 a)

Synthesis of title compound was performed as described for rac-4 b starting from 3 a (100.0 mg), rac-4 a was obtained as a white solid (90.1 mg, 90%). NMR spectra were indistinguishable from that of S-4 a.
Steady-State Kinetic Studies of DpkAPsyrin for Sodium Pyruvate
The kinetic parameters for DpkAPsyrin were determined in a continuous assay method monitoring the oxidation of NADPH to NADP+ at 340 nm (NADPH ϵ340=6.22 mM−1 cm−1) using sodium pyruvate as substrates (Figure S10A). The reaction was monitored during 15 min measuring each 30 s. The assay mixture (0.3 mL) consisted of 50 mM Tris-HCl buffer pH 8.0, containing NADPH (0.16 mM), sodium pyruvate (1–60 mM) and appropriate amounts of enzymes (i. e., optimal range of enzyme concentration determined for each substrate, Figure S10B). One unit of activity was defined as the amount of enzyme that catalyze the formation of 1 μmol NADP+ per min at 30 °C. Measurements were carried out in triplicate independent experiments. To determine the kinetic parameters, data were fitted to the Michaelis-Menten kinetic model using the software GraphPad Prism version 5.0 (Figure S10C).
X-Rays
CCDC-2250740, 2250741, and 2250742 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/structures).
Acknowledgments
Grant PID2021-122166OB−I00 funded by MCIN/AEI/10.13039/501100011033, and by “ERDF A way of making Europe”. CJM acknowledges a PhD contract (i. e. Ayudas para Contratos Predoctorales para la Formación de Doctores) BES-2016-079447 funded by MCIN/AEI/10.13039/501100011033. The authors thankfully acknowledge the use of the computational resources of the Consorci de Serveis Universitaris de Catalunya (CSUC).
中文翻译:
扩大丁香假单胞菌 Δ1-哌啶-2-羧酸盐/Δ1-吡咯啉-2-羧酸盐还原酶 (DpkAPsyrin) 的合成应用。(S,E)-2-羟基-4-芳基丁-3-烯酸衍生物的生物催化不对称合成
手性2-羟基-4-芳基丁-3-烯酸衍生物是合成血管紧张素转换酶(ACE)抑制剂的重要前体,例如依那普利、赖诺普利、西拉普利或贝那普利。在这项工作中,我们利用来自丁香假单胞菌的 Δ 1 -哌啶-2-羧酸盐/Δ 1 -吡咯啉-2-羧酸盐还原酶的未探索的混杂酮还原酶活性。番茄 DSM 50315 (DpkA Psyrin ) 用于合成 ( S , E )-2-羟基-4-芳基丁-3-烯酸。该策略被设计为酶级联,包括丙酮酸与芳基醛之间的羟醛缩合,由反式催化来自恶臭假单胞菌(HBPA Pputida )的 -o-羟基亚苄基丙酮酸水合酶-醛缩酶,用于构建碳支架,以及随后由 DpkA Psyrin催化的羰基的不对称还原。酶级联提供了定量转化,总体分离产量在 57-85% 之间。总共制备了九种结构不同的 ( S , E )-2-羟基-4-芳基丁-3-烯酸,ee 含量在 87-99% 之间。































 京公网安备 11010802027423号
京公网安备 11010802027423号