当前位置:
X-MOL 学术
›
J. Phys. Chem. Lett.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Density-Corrected Density Functional Theory for Molecular Properties
The Journal of Physical Chemistry Letters ( IF 4.8 ) Pub Date : 2023-05-23 , DOI: 10.1021/acs.jpclett.3c00953 Pierpaolo Morgante 1 , Jochen Autschbach 1
The Journal of Physical Chemistry Letters ( IF 4.8 ) Pub Date : 2023-05-23 , DOI: 10.1021/acs.jpclett.3c00953 Pierpaolo Morgante 1 , Jochen Autschbach 1
Affiliation
|
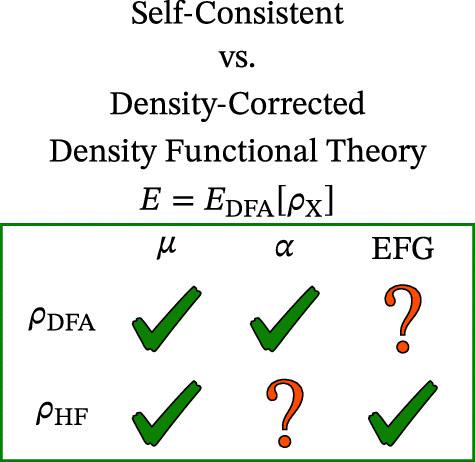
Density-corrected (DC) density functional theory (DFT) has been proposed to overcome difficulties related to the self-interaction error. The procedure uses the Hartree–Fock electron density (matrix) non-self-consistently in conjunction with an approximate functional. DC-DFT has so far mainly been tested for total energy differences, whereas other types of molecular properties have not been evaluated systematically. This work focuses on the performance of DC-DFT for molecular properties, namely, dipole moments, static polarizabilities, and electric field gradients (EFGs) at atomic nuclei. Accurate reference data were generated from coupled-cluster theory to assess the performance of DC and self-consistent DFT calculations for twelve molecules, including diatomics with transition metals. DC-DFT does no harm in dipole moment calculations, but it negatively impacts the polarizability in at least one case. DC-DFT performs well for EFGs, even for the difficult case of CuCl.
中文翻译:

分子性质的密度校正密度泛函理论
已经提出密度校正(DC)密度泛函理论(DFT)来克服与自相互作用误差相关的困难。该过程结合近似泛函非自洽地使用 Hartree–Fock 电子密度(矩阵)。到目前为止,DC-DFT 主要针对总能量差异进行了测试,而其他类型的分子特性尚未得到系统评估。这项工作的重点是 DC-DFT 的分子特性性能,即偶极矩、静态极化率和原子核处的电场梯度 (EFG)。准确的参考数据是从耦合簇理论生成的,用于评估 12 个分子(包括具有过渡金属的双原子)的 DC 和自洽 DFT 计算的性能。DC-DFT 对偶极矩计算无害,但它至少在一种情况下会对极化率产生负面影响。DC-DFT 对于 EFG 表现良好,即使对于 CuCl 的困难情况也是如此。
更新日期:2023-05-23
中文翻译:
分子性质的密度校正密度泛函理论
已经提出密度校正(DC)密度泛函理论(DFT)来克服与自相互作用误差相关的困难。该过程结合近似泛函非自洽地使用 Hartree–Fock 电子密度(矩阵)。到目前为止,DC-DFT 主要针对总能量差异进行了测试,而其他类型的分子特性尚未得到系统评估。这项工作的重点是 DC-DFT 的分子特性性能,即偶极矩、静态极化率和原子核处的电场梯度 (EFG)。准确的参考数据是从耦合簇理论生成的,用于评估 12 个分子(包括具有过渡金属的双原子)的 DC 和自洽 DFT 计算的性能。DC-DFT 对偶极矩计算无害,但它至少在一种情况下会对极化率产生负面影响。DC-DFT 对于 EFG 表现良好,即使对于 CuCl 的困难情况也是如此。









































 京公网安备 11010802027423号
京公网安备 11010802027423号