Nature Genetics ( IF 31.7 ) Pub Date : 2023-05-11 , DOI: 10.1038/s41588-023-01392-0 Zikun Yang 1 , Chen Wang 1, 2 , Linxi Liu 3 , Atlas Khan 2 , Annie Lee 4 , Badri Vardarajan 4 , Richard Mayeux 4 , Krzysztof Kiryluk 2 , Iuliana Ionita-Laza 1
|
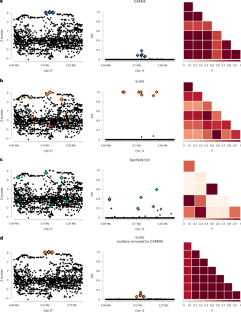
Fine-mapping is commonly used to identify putative causal variants at genome-wide significant loci. Here we propose a Bayesian model for fine-mapping that has several advantages over existing methods, including flexible specification of the prior distribution of effect sizes, joint modeling of summary statistics and functional annotations and accounting for discrepancies between summary statistics and external linkage disequilibrium in meta-analyses. Using simulations, we compare performance with commonly used fine-mapping methods and show that the proposed model has higher power and lower false discovery rate (FDR) when including functional annotations, and higher power, lower FDR and higher coverage for credible sets in meta-analyses. We further illustrate our approach by applying it to a meta-analysis of Alzheimer’s disease genome-wide association studies where we prioritize putatively causal variants and genes.
中文翻译:
CARMA 是一种新的贝叶斯模型,用于全基因组关联荟萃分析中的精细映射
精细作图通常用于识别全基因组重要位点的假定因果变异。在这里,我们提出了一种用于精细映射的贝叶斯模型,它比现有方法有几个优点,包括效应大小先验分布的灵活规范、汇总统计和功能注释的联合建模以及解释汇总统计和元中外部链接不平衡之间的差异。 - 分析。通过模拟,我们将性能与常用的精细映射方法进行了比较,并表明所提出的模型在包含功能注释时具有更高的功效和更低的错误发现率(FDR),并且对于元中的可信集具有更高的功效、更低的FDR和更高的覆盖率。分析。我们通过将其应用于阿尔茨海默病全基因组关联研究的荟萃分析来进一步说明我们的方法,其中我们优先考虑假定的因果变异和基因。










































 京公网安备 11010802027423号
京公网安备 11010802027423号