Chemical Physics ( IF 2.0 ) Pub Date : 2023-03-21 , DOI: 10.1016/j.chemphys.2023.111891 Kaifu Zhong , Xinghong Cai , Min Wang
|
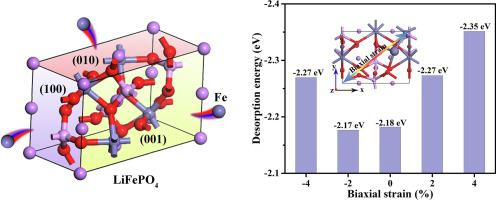
Different surface models are built to explain the desorption properties of Fe atoms in LiFePO4 and FePO4 structures based on density functional theory (DFT) computations. Analyses of the band structures describe the pristine LiFePO4 and FePO4 structures with semiconducting (0.67 eV) and metallic properties, respectively. The electronic properties of the different surfaces mainly arise from the contributions of O and Fe atoms. Calculations of desorption energies and electronic properties reveal that the (1 0 0) surfaces of LiFePO4 and FePO4 are most favourable for desorption of Fe atoms, while the (0 1 0) surface is less susceptible. In addition, the investigations of Fe atom desorption energy at different strains indicate that the applications of strain increase the tendency of Fe’s desorption on LiFePO4 (1 0 0) surface. This study presents a theoretical foundation for understanding the correlation between different surfaces and desorption of Fe atoms for the future applications of lithium ion battery.
中文翻译:
铁原子在LiFePO4和FePO4的(1 0 0)表面更易脱附的机理
基于密度泛函理论 (DFT) 计算,建立了不同的表面模型来解释 LiFePO 4和 FePO 4结构中 Fe 原子的解吸特性。能带结构分析描述了分别具有半导体 (0.67 eV) 和金属特性的原始 LiFePO 4和 FePO 4结构。不同表面的电子特性主要来自O和Fe原子的贡献。解吸能和电子性质的计算表明,LiFePO 4和FePO 4的(1 0 0)表面最有利于Fe原子的解吸,而(0 1 0) 表面不易受影响。此外,对不同应变下Fe原子解吸能的研究表明,应变的施加增加了Fe在LiFePO 4 (1 0 0)表面的解吸趋势。该研究为了解不同表面与铁原子解吸之间的相关性,为锂离子电池的未来应用提供了理论基础。










































 京公网安备 11010802027423号
京公网安备 11010802027423号