当前位置:
X-MOL 学术
›
Adv. Theory Simul.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Computing the Lattice Thermal Conductivity of Small-Molecule Organic Semiconductors: A Systematic Comparison of Molecular Dynamics Based Methods
Advanced Theory and Simulations ( IF 2.9 ) Pub Date : 2023-03-08 , DOI: 10.1002/adts.202200892 Alexandre Vercouter 1 , Vincent Lemaur 1 , Claudio Melis 2 , Jérôme Cornil 1
Advanced Theory and Simulations ( IF 2.9 ) Pub Date : 2023-03-08 , DOI: 10.1002/adts.202200892 Alexandre Vercouter 1 , Vincent Lemaur 1 , Claudio Melis 2 , Jérôme Cornil 1
Affiliation
|
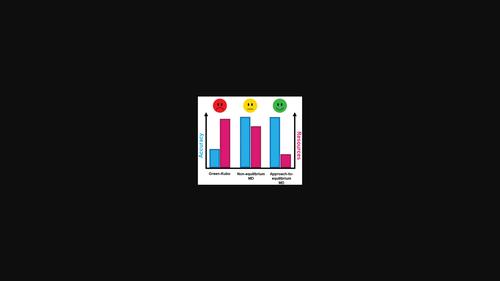
While the Green-Kubo and non-equilibrium molecular dynamics methods have been compared quite extensively to calculate the thermal conductivity in inorganic compounds, there is currently a lack of comparison of these algorithms with the more recently developed approach-to-equilibrium molecular dynamics (AEMD) method for other types of materials such as organic semiconductors. To fill this gap, this article reports a theoretical description of thermal transport in single crystals made of terthiophene as prototypical system based on the three most popular molecular dynamics approaches. A systematic comparison of the computed values of thermal conductivity and its anisotropy is carried out and the strengths and weaknesses associated with each method are discussed. Although the three algorithms give essentially the same trends, this study points to the “AEMD” approach as the most suitable compromise between accuracy and computing cost. On the material aspects, the theoretical modeling yields an anisotropic character of the thermal transport in crystals whose out-of-plane thermal conductivity component is approximately twice larger than the in-plane components. The AEMD approach is further used to investigate the influence of temperature on thermal transport in terthiophene. The trends are utterly rationalized by relying on the concepts of phonon mean free paths and phonon group velocities.
中文翻译:

计算小分子有机半导体的晶格热导率:基于分子动力学方法的系统比较
虽然已经对 Green-Kubo 和非平衡分子动力学方法进行了相当广泛的比较,以计算无机化合物的热导率,但目前缺乏将这些算法与最近开发的平衡分子动力学方法 (AEMD) 进行比较) 方法用于其他类型的材料,如有机半导体。为了填补这一空白,本文基于三种最流行的分子动力学方法,报告了对由三联噻吩制成的单晶热传输作为原型系统的理论描述。对导热系数的计算值及其各向异性进行了系统比较,并讨论了每种方法的优缺点。尽管这三种算法给出了基本相同的趋势,该研究指出“AEMD”方法是准确性和计算成本之间最合适的折衷方案。在材料方面,理论建模产生了晶体中热传输的各向异性特征,其面外热导率分量大约是面内分量的两倍。AEMD 方法进一步用于研究温度对三联噻吩热传输的影响。依靠声子平均自由程和声子群速度的概念,这些趋势完全合理化了。理论模型产生了晶体中热传输的各向异性特征,其面外热导率分量大约是面内分量的两倍。AEMD 方法进一步用于研究温度对三联噻吩热传输的影响。依靠声子平均自由程和声子群速度的概念,这些趋势完全合理化了。理论模型产生了晶体中热传输的各向异性特征,其面外热导率分量大约是面内分量的两倍。AEMD 方法进一步用于研究温度对三联噻吩热传输的影响。依靠声子平均自由程和声子群速度的概念,这些趋势完全合理化了。
更新日期:2023-03-08
中文翻译:
计算小分子有机半导体的晶格热导率:基于分子动力学方法的系统比较
虽然已经对 Green-Kubo 和非平衡分子动力学方法进行了相当广泛的比较,以计算无机化合物的热导率,但目前缺乏将这些算法与最近开发的平衡分子动力学方法 (AEMD) 进行比较) 方法用于其他类型的材料,如有机半导体。为了填补这一空白,本文基于三种最流行的分子动力学方法,报告了对由三联噻吩制成的单晶热传输作为原型系统的理论描述。对导热系数的计算值及其各向异性进行了系统比较,并讨论了每种方法的优缺点。尽管这三种算法给出了基本相同的趋势,该研究指出“AEMD”方法是准确性和计算成本之间最合适的折衷方案。在材料方面,理论建模产生了晶体中热传输的各向异性特征,其面外热导率分量大约是面内分量的两倍。AEMD 方法进一步用于研究温度对三联噻吩热传输的影响。依靠声子平均自由程和声子群速度的概念,这些趋势完全合理化了。理论模型产生了晶体中热传输的各向异性特征,其面外热导率分量大约是面内分量的两倍。AEMD 方法进一步用于研究温度对三联噻吩热传输的影响。依靠声子平均自由程和声子群速度的概念,这些趋势完全合理化了。理论模型产生了晶体中热传输的各向异性特征,其面外热导率分量大约是面内分量的两倍。AEMD 方法进一步用于研究温度对三联噻吩热传输的影响。依靠声子平均自由程和声子群速度的概念,这些趋势完全合理化了。










































 京公网安备 11010802027423号
京公网安备 11010802027423号