Analytica Chimica Acta ( IF 5.7 ) Pub Date : 2023-03-02 , DOI: 10.1016/j.aca.2023.341038 Wei Fang 1 , Zhuokun Du 1 , Linlin Kong 1 , Bin Fu 1 , Guibin Wang 1 , Yangjun Zhang 1 , Weijie Qin 2
|
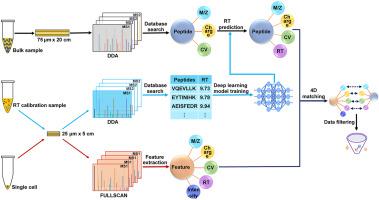
Single-cell analysis has received much attention in recent years for elucidating the widely existing cellular heterogeneity in biological systems. However, the ability to measure the proteome in single cells is still far behind that of transcriptomics due to the lack of sensitive and high-throughput mass spectrometry methods. Herein, we report an integrated strategy termed “SCP-MS1” that combines fast liquid chromatography (LC) separation, deep learning-based retention time (RT) prediction and MS1-only acquisition for rapid and sensitive single-cell proteome analysis. In SCP-MS1, the peptides were identified via four-dimensional MS1 feature (m/z, RT, charge and FAIMS CV) matching, therefore relieving MS acquisition from the time consuming and information losing MS2 step and making this method particularly compatible with fast LC separation. By completely omitting the MS2 step, all the MS analysis time was utilized for MS1 acquisition in SCP-MS1 and therefore led to 65%–138% increased MS1 feature collection. Unlike “match between run” methods that still needed MS2 information for RT alignment, SCP-MS1 used deep learning-based RT prediction to transfer the measured RTs in long gradient bulk analyses to short gradient single cell analyses, which was the key step to enhance both identification scale and matching accuracy. Using this strategy, more than 2000 proteins were obtained from 0.2 ng of peptides with a 14-min active gradient at a false discovery rate (FDR) of 0.8%. Comparing with the DDA method, improved quantitative performance was also observed for SCP-MS1 with approximately 50% decreased median coefficient of variation of quantified proteins. For single-cell analysis, 1715 ± 204 and 1604 ± 224 proteins were quantified in single 293T and HeLa cells, respectively. Finally, SCP-MS1 was applied to single-cell proteome analysis of sorafenib resistant and non-resistant HepG2 cells and revealed clear cellular heterogeneity in the resistant population that may be masked in bulk studies.
中文翻译:
基于快速液相色谱分离、保留时间预测和仅 MS1 采集的快速灵敏的单细胞蛋白质组学方法
近年来,单细胞分析因阐明生物系统中广泛存在的细胞异质性而备受关注。然而,由于缺乏灵敏和高通量的质谱方法,测量单细胞蛋白质组的能力仍然远远落后于转录组学。在此,我们报告了一种称为“SCP-MS1”的综合策略,它结合了快速液相色谱 (LC) 分离、基于深度学习的保留时间 (RT) 预测和仅 MS1 采集,用于快速和灵敏的单细胞蛋白质组分析。在 SCP-MS1 中,肽是通过四维 MS1 特征 ( m / z, RT, charge 和 FAIMS CV) 匹配,因此将 MS 采集从耗时且信息丢失的 MS2 步骤中解脱出来,并使该方法特别适用于快速 LC 分离。通过完全省略 MS2 步骤,所有 MS 分析时间都用于 SCP-MS1 中的 MS1 采集,因此导致 MS1 特征收集增加 65%–138%。与仍然需要 MS2 信息进行 RT 对齐的“运行之间的匹配”方法不同,SCP-MS1 使用基于深度学习的 RT 预测将长梯度批量分析中测量的 RT 转移到短梯度单细胞分析中,这是增强的关键步骤识别尺度和匹配精度。使用此策略,以 0.8% 的错误发现率 (FDR) 以 14 分钟的活性梯度从 0.2 ng 肽中获得了 2000 多种蛋白质。与 DDA 方法相比,SCP-MS1 的定量性能也有所提高,量化蛋白质的中值变异系数降低了约 50%。对于单细胞分析,分别在单个 293T 和 HeLa 细胞中定量了 1715 ± 204 和 1604 ± 224 种蛋白质。最后,将 SCP-MS1 应用于索拉非尼耐药和非耐药 HepG2 细胞的单细胞蛋白质组分析,并揭示了耐药群体中明显的细胞异质性,这可能在大量研究中被掩盖。










































 京公网安备 11010802027423号
京公网安备 11010802027423号