当前位置:
X-MOL 学术
›
Acc. Chem. Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Sulfoxide-Directed or 3d-Metal Catalyzed C–H Activation and Hypervalent Iodines as Tools for Atroposelective Synthesis
Accounts of Chemical Research ( IF 16.4 ) Pub Date : 2023-01-27 , DOI: 10.1021/acs.accounts.2c00573 Sabine Choppin 1 , Joanna Wencel-Delord 1
Accounts of Chemical Research ( IF 16.4 ) Pub Date : 2023-01-27 , DOI: 10.1021/acs.accounts.2c00573 Sabine Choppin 1 , Joanna Wencel-Delord 1
Affiliation
|
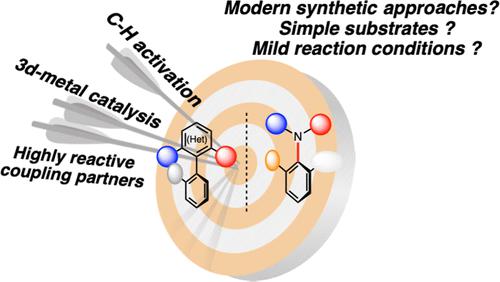
The expanding applications of atropisomeric compounds combined with the growing diversity of such chiral molecules translate into an urgent need for innovative synthetic strategies allowing their rapid, efficient, and sustainable synthesis. Recently, the C–H activation approach has provided new opportunities for synthesizing axially chiral compounds. The two complementary approaches allowing implementation of the C–H activation methodology toward the synthesis of the chiral molecules imply either ortho-functionalization of the preexisting prochiral or atropo-unstable biaryl substrates or direct C–H arylation of sterically encumbered aromatics. The first approach required the preinstallation of a directing group on a biaryl precursor, which drastically limits the diversity of thus generated products. To tackle this important synthetic limitation, we have envisioned using a chiral sulfoxide as both directing group and chiral auxiliary. Indeed, in addition to efficiently coordinating the Pd-catalyst thus allowing chiral induction, the sulfoxide moiety can be easily removed, via the sulfoxide/lithium exchange, after the C–H activation step, thus guaranteeing an almost unlimited postdiversification of the atropisomeric products. The efficiency and generality of this concept could be illustrated by developing atropo-diastereoselective oxidative Heck reaction, direct acetoxylation, and iodination, as well as direct arylation. Besides, the synthetic utility of this methodology was demonstrated by designing an expedient synthesis of a direct steganone precursor. This unique transformation also allowed us to build up unprecedented triaryl scaffolds with two perfectly controlled chiral axes, original chiral skeletons for new ligand design. While considering the atroposelective direct arylations, the clear antagonism between the harsh reaction conditions frequently required for the coupling of two sterically hindered compounds and the atropo-stability of the new product, resulted in the scarcity of such transformations. To solve this fundamental challenge, we have focused on the application of a low-valent cobalt catalyst, prompted to catalyze C–H activation of indoles at the C2 position under extremely mild reaction conditions (room temperature). Accordingly, atroposelective C2-arylation of indoles could be achieved using an original carbene ligand and delivering the uncommon atropoisomerically pure indoles in excellent yields and enantioselectivities. Detailed combined experimental and theoretical mechanistic studies shed light on the mechanism of this transformation, providing strong evidence regarding the origin of the enantioselectivity. Finally, the antagonism between steric hindrance required to guarantee the atropo-stability of a molecule and harsh reaction conditions required to couple two partners is a strong limitation not only for the development of atroposelective C–H arylation reaction but also for the development of direct synthesis of the C–N axially chiral compounds. Despite the long history and incredible advances achieved in Ullmann–Goldberg and Buchwald–Hartwig couplings, atroposelective versions of such transformations have remained unprecedented until recently. Our idea to tackle this challenging issue consisted in using hypervalent iodines as highly reactive coupling partners, thus allowing the desired N-arylations to occur at room temperature. This hypothesis could be validated by reporting first atropo-diastereoselective Cu-catalyzed N-arylation, using sulfoxide λ3-iodanes as the coupling partners. Subsequently, the enantioselective version of this atroposelective N-arylation was successfully established by using a chiral Cu-complex bearing a BOX ligand. In conclusion, we report herein designing tailored-made solutions to provide new synthetic strategies to construct the atropisomeric molecules, including biaryls and C–N axially chiral molecules.
中文翻译:

亚砜导向或 3d 金属催化的 C-H 活化和高价碘作为阻转选择性合成的工具
阻转异构化合物的应用范围不断扩大,加上此类手性分子的多样性不断增加,因此迫切需要创新的合成策略,以实现快速、高效和可持续的合成。最近,C-H 活化方法为合成轴向手性化合物提供了新的机会。这两种互补的方法允许对手性分子的合成实施 C-H 活化方法,这意味着要么对预先存在的前手性或 atropo 不稳定的联芳基底物进行邻位官能化,要么对空间阻碍的芳烃进行直接 C-H 芳基化。第一种方法需要在联芳基前体上预先安装一个导向基团,这极大地限制了由此产生的产品的多样性。为了解决这个重要的合成限制,我们设想使用手性亚砜作为导向基团和手性助剂。事实上,除了有效协调 Pd 催化剂从而允许手性诱导外,在 C-H 活化步骤后,亚砜部分可以通过亚砜/锂交换轻松去除,从而保证阻转异构产物几乎无限的后多样化。这一概念的有效性和普遍性可以通过发展atropo-diastereoselective 氧化Heck 反应、直接乙酰氧基化、碘化以及直接芳基化来说明。此外,该方法的合成效用通过设计一种直接的 steganone 前体的便捷合成得到证明。这种独特的转变还使我们能够构建具有两个完美控制的手性轴的前所未有的三芳基支架,用于新配体设计的原始手性骨架。在考虑阻转选择性直接芳基化时,两种空间位阻化合物偶联经常需要的苛刻反应条件与新产品的阻转稳定性之间存在明显的拮抗作用,导致此类转化的稀缺性。为了解决这一根本性挑战,我们专注于低价钴催化剂的应用,促使其在极其温和的反应条件(室温)下催化 C2 位吲哚的 C-H 活化。因此,使用原始卡宾配体可以实现吲哚的阻转选择性 C2-芳基化,并以优异的收率和对映选择性提供不常见的阻转异构纯吲哚。详细的实验和理论机制相结合的研究揭示了这种转变的机制,为对映选择性的起源提供了强有力的证据。最后,保证分子的阻转稳定性所需的空间位阻与偶联两个伙伴所需的苛刻反应条件之间的拮抗作用不仅严重限制了阻转选择性 C-H 芳基化反应的发展,而且严重限制了直接合成的发展C-N 轴向手性化合物。尽管 Ullmann-Goldberg 和 Buchwald-Hartwig 耦合具有悠久的历史并取得了令人难以置信的进步,但直到最近,这种转变的逆选择性版本仍然是前所未有的。我们解决这一具有挑战性问题的想法包括使用高价碘作为高反应性偶联伙伴,从而允许在室温下发生所需的 N-芳基化。该假设可以通过使用亚砜 λ 报告第一个 atropo-diastereoselective Cu 催化的 N-芳基化来验证3 -碘作为偶联伙伴。随后,通过使用带有 BOX 配体的手性 Cu 络合物,成功地建立了这种阻转选择性 N-芳基化的对映选择性版本。总之,我们在此报告了设计量身定制的解决方案,以提供新的合成策略来构建阻转异构分子,包括联芳基和 C-N 轴向手性分子。
更新日期:2023-01-27
中文翻译:
亚砜导向或 3d 金属催化的 C-H 活化和高价碘作为阻转选择性合成的工具
阻转异构化合物的应用范围不断扩大,加上此类手性分子的多样性不断增加,因此迫切需要创新的合成策略,以实现快速、高效和可持续的合成。最近,C-H 活化方法为合成轴向手性化合物提供了新的机会。这两种互补的方法允许对手性分子的合成实施 C-H 活化方法,这意味着要么对预先存在的前手性或 atropo 不稳定的联芳基底物进行邻位官能化,要么对空间阻碍的芳烃进行直接 C-H 芳基化。第一种方法需要在联芳基前体上预先安装一个导向基团,这极大地限制了由此产生的产品的多样性。为了解决这个重要的合成限制,我们设想使用手性亚砜作为导向基团和手性助剂。事实上,除了有效协调 Pd 催化剂从而允许手性诱导外,在 C-H 活化步骤后,亚砜部分可以通过亚砜/锂交换轻松去除,从而保证阻转异构产物几乎无限的后多样化。这一概念的有效性和普遍性可以通过发展atropo-diastereoselective 氧化Heck 反应、直接乙酰氧基化、碘化以及直接芳基化来说明。此外,该方法的合成效用通过设计一种直接的 steganone 前体的便捷合成得到证明。这种独特的转变还使我们能够构建具有两个完美控制的手性轴的前所未有的三芳基支架,用于新配体设计的原始手性骨架。在考虑阻转选择性直接芳基化时,两种空间位阻化合物偶联经常需要的苛刻反应条件与新产品的阻转稳定性之间存在明显的拮抗作用,导致此类转化的稀缺性。为了解决这一根本性挑战,我们专注于低价钴催化剂的应用,促使其在极其温和的反应条件(室温)下催化 C2 位吲哚的 C-H 活化。因此,使用原始卡宾配体可以实现吲哚的阻转选择性 C2-芳基化,并以优异的收率和对映选择性提供不常见的阻转异构纯吲哚。详细的实验和理论机制相结合的研究揭示了这种转变的机制,为对映选择性的起源提供了强有力的证据。最后,保证分子的阻转稳定性所需的空间位阻与偶联两个伙伴所需的苛刻反应条件之间的拮抗作用不仅严重限制了阻转选择性 C-H 芳基化反应的发展,而且严重限制了直接合成的发展C-N 轴向手性化合物。尽管 Ullmann-Goldberg 和 Buchwald-Hartwig 耦合具有悠久的历史并取得了令人难以置信的进步,但直到最近,这种转变的逆选择性版本仍然是前所未有的。我们解决这一具有挑战性问题的想法包括使用高价碘作为高反应性偶联伙伴,从而允许在室温下发生所需的 N-芳基化。该假设可以通过使用亚砜 λ 报告第一个 atropo-diastereoselective Cu 催化的 N-芳基化来验证3 -碘作为偶联伙伴。随后,通过使用带有 BOX 配体的手性 Cu 络合物,成功地建立了这种阻转选择性 N-芳基化的对映选择性版本。总之,我们在此报告了设计量身定制的解决方案,以提供新的合成策略来构建阻转异构分子,包括联芳基和 C-N 轴向手性分子。








































 京公网安备 11010802027423号
京公网安备 11010802027423号