Journal of Fluorescence ( IF 2.6 ) Pub Date : 2022-12-01 , DOI: 10.1007/s10895-022-03077-z Said Zouitina 1 , Abdelkhalk Aboulouard 2, 3 , Ahlam El Ghazali 1 , Abdessamad Tounsi 1 , Mohammed El Idrissi 4
|
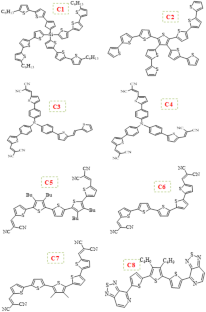
In this research work, we study the structural, optical, electronic, and photovoltaic properties of eight thiophene-based π-conjugated organic molecules using quantum methods namely time-dependent density functional theory. In particular, we identify the relationships between the chemical structure of these π-conjugated organic molecules and their optoelectronic properties. Moreover, we calculate and compare the highest energy occupied molecular orbital and lowest energy unoccupied molecular orbital energy levels of these compounds which act as donor with the ones of the acceptorphenyl-C61-butyric acid methyl ester. As a result, the investigated molecules show a low band gap, suitable open-circuit voltage and appropriate alignment energy level between the engineered donor molecules and the acceptor phenyl-C61-butyric acid methyl ester. This theoretical study shows that these new molecules have potential properties for the development of organic heterojunction photovoltaic cells.
中文翻译:
基于噻吩的新型小分子作为体异质结光伏电池供体材料的计算研究
在这项研究工作中,我们使用量子方法(即时间相关密度泛函理论)研究了八种基于噻吩的 π- 共轭有机分子的结构、光学、电子和光伏特性。特别是,我们确定了这些 π 共轭有机分子的化学结构与其光电特性之间的关系。此外,我们计算并比较了这些作为供体的化合物与受体苯基-C 61 -丁酸甲酯的最高能量占据分子轨道和最低能量未占据分子轨道能级。因此,所研究的分子在工程供体分子和受体苯基-C 之间显示出低带隙、合适的开路电压和合适的排列能级61-丁酸甲酯。该理论研究表明,这些新分子具有开发有机异质结光伏电池的潜在特性。










































 京公网安备 11010802027423号
京公网安备 11010802027423号