当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Photochemical carbon–sulfur bond cleavage in thioethers mediated via excited state Rydberg-to-valence evolution
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2022-12-01 , DOI: 10.1039/d2cp04789f Nikoleta Kotsina 1 , Sebastian L Jackson 1 , Thomas Malcomson 2 , Martin J Paterson 3 , Dave Townsend 1, 3
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2022-12-01 , DOI: 10.1039/d2cp04789f Nikoleta Kotsina 1 , Sebastian L Jackson 1 , Thomas Malcomson 2 , Martin J Paterson 3 , Dave Townsend 1, 3
Affiliation
|
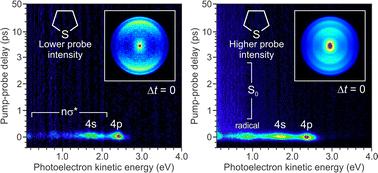
Time-resolved photoelectron imaging and supporting ab initio quantum chemistry calculations were used to investigate non-adiabatic excess energy redistribution dynamics operating in the saturated thioethers diethylsulfide, tetrahydrothiophene and thietane. In all cases, 200 nm excitation leads to molecular fragmentation on an ultrafast (<100 fs) timescale, driven by the evolution of Rydberg-to-valence orbital character along the S–C stretching coordinate. The C–S–C bending angle was also found to be a key coordinate driving initial internal conversion through the excited state Rydberg manifold, although only small angular displacements away from the ground state equilibrium geometry are required. Conformational constraints imposed by the cyclic ring structures of tetrahydrothiophene and thietane do not therefore influence dynamical timescales to any significant extent. Through use of a high-intensity 267 nm probe, we were also able to detect the presence of some transient (bi)radical species. These are extremely short lived, but they appear to confirm the presence of two competing excited state fragmentation channels – one proceeding directly from the initially prepared 4p manifold, and one involving non-adiabatic population of the 4s state. This is in addition to a decay pathway leading back to the S0 electronic ground state, which shows an enhanced propensity in the 5-membered ring system tetrahydrothiophene over the other two species investigated.
中文翻译:

硫醚中的光化学碳硫键断裂通过激发态里德堡到价的演化介导
时间分辨光电子成像和支持从头算量子化学计算用于研究在饱和硫醚二乙基硫醚、四氢噻吩和硫杂环丁烷中运行的非绝热过剩能量再分配动力学。在所有情况下,200 nm 激发都会导致超快(<100 fs)时间尺度上的分子碎裂,这是由 Rydberg-to-valence 轨道特征沿 S-C 伸缩坐标的演变驱动的。C-S-C 弯曲角也被发现是通过激发态里德堡流形驱动初始内部转换的关键坐标,尽管只需要远离基态平衡几何的小角位移。因此,四氢噻吩和硫杂环丁烷的环状环结构施加的构象限制不会在任何显着程度上影响动力学时间尺度。通过使用高强度 267 nm 探针,我们还能够检测到一些瞬态(双)自由基物种的存在。这些寿命极短,但它们似乎证实了两个相互竞争的激发态碎裂通道的存在——一个直接从最初准备好的 4p 流形开始,另一个涉及 4s 状态的非绝热布居。这是通往 S 的衰变途径的补充0电子基态,这表明在五元环系统四氢噻吩中比其他两个研究的物种具有增强的倾向。
更新日期:2022-12-01
中文翻译:
硫醚中的光化学碳硫键断裂通过激发态里德堡到价的演化介导
时间分辨光电子成像和支持从头算量子化学计算用于研究在饱和硫醚二乙基硫醚、四氢噻吩和硫杂环丁烷中运行的非绝热过剩能量再分配动力学。在所有情况下,200 nm 激发都会导致超快(<100 fs)时间尺度上的分子碎裂,这是由 Rydberg-to-valence 轨道特征沿 S-C 伸缩坐标的演变驱动的。C-S-C 弯曲角也被发现是通过激发态里德堡流形驱动初始内部转换的关键坐标,尽管只需要远离基态平衡几何的小角位移。因此,四氢噻吩和硫杂环丁烷的环状环结构施加的构象限制不会在任何显着程度上影响动力学时间尺度。通过使用高强度 267 nm 探针,我们还能够检测到一些瞬态(双)自由基物种的存在。这些寿命极短,但它们似乎证实了两个相互竞争的激发态碎裂通道的存在——一个直接从最初准备好的 4p 流形开始,另一个涉及 4s 状态的非绝热布居。这是通往 S 的衰变途径的补充0电子基态,这表明在五元环系统四氢噻吩中比其他两个研究的物种具有增强的倾向。










































 京公网安备 11010802027423号
京公网安备 11010802027423号