Computational and Structural Biotechnology Journal ( IF 6 ) Pub Date : 2022-11-25 , DOI: 10.1016/j.csbj.2022.11.048 Audrone Valanciute 1 , Lasse Nygaard 1 , Henrike Zschach 2 , Michael Maglegaard Jepsen 1 , Kresten Lindorff-Larsen 1 , Amelie Stein 2
|
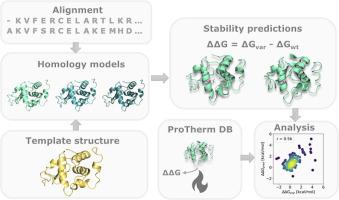
Calculating changes in protein stability (ΔΔG) has been shown to be central for predicting the consequences of single amino acid substitutions in protein engineering as well as interpretation of genomic variants for disease risk. Structure-based calculations are considered most accurate, however the tools used to calculate ΔΔGs have been developed on experimentally resolved structures. Extending those calculations to homology models based on related proteins would greatly extend their applicability as large parts of e.g. the human proteome are not structurally resolved. In this study we aim to investigate the accuracy of ΔΔG values predicted on homology models compared to crystal structures. Specifically, we identified four proteins with a large number of experimentally tested ΔΔGs and templates for homology modeling across a broad range of sequence identities, and selected three methods for ΔΔG calculations to test. We find that ΔΔG-values predicted from homology models compare equally well to experimental ΔΔGs as those predicted on experimentally established crystal structures, as long as the sequence identity of the model template to the target protein is at least 40%. In particular, the Rosetta cartesian_ddg protocol is robust against the small perturbations in the structure which homology modeling introduces. In an independent assessment, we observe a similar trend when using ΔΔGs to categorize variants as low or wild-type-like abundance. Overall, our results show that stability calculations performed on homology models can substitute for those on crystal structures with acceptable accuracy as long as the model is built on a template with sequence identity of at least 40% to the target protein.
中文翻译:
从同源模型准确预测蛋白质稳定性
计算蛋白质稳定性的变化 (ΔΔG) 已被证明对于预测蛋白质工程中单个氨基酸取代的后果以及解释基因组变异对疾病风险的影响至关重要。基于结构的计算被认为是最准确的,但是用于计算 ΔΔGs 的工具是在实验解析的结构上开发的。将这些计算扩展到基于相关蛋白质的同源模型将极大地扩展它们的适用性,因为例如人类蛋白质组的大部分在结构上没有解析。在这项研究中,我们旨在研究与晶体结构相比,在同源模型上预测的 ΔΔG 值的准确性。具体来说,我们确定了四种蛋白质,它们具有大量经过实验测试的 ΔΔG 和模板,用于在广泛的序列同一性范围内进行同源建模,并选择了三种 ΔΔG 计算方法进行测试。我们发现,只要模型模板与靶蛋白的序列同一性至少为 40%,从同源模型预测的 ΔΔG 值与实验性 ΔΔGs 相比,与在实验建立的晶体结构上预测的值同样好。特别是,罗塞塔cartesian_ddg协议对同源建模引入的结构中的小扰动具有鲁棒性。在一项独立评估中,我们在使用 ΔΔGs 将变异分类为低丰度或类野生型丰度时观察到类似的趋势。总的来说,我们的结果表明,只要模型建立在与目标蛋白质具有至少 40% 的序列同一性的模板上,在同源模型上进行的稳定性计算就可以以可接受的精度替代晶体结构上的稳定性计算。


























 京公网安备 11010802027423号
京公网安备 11010802027423号