当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Spin-State Splittings in 3d Transition-Metal Complexes Revisited: Benchmarking Approximate Methods for Adiabatic Spin-State Energy Differences in Fe(II) Complexes
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2022-11-23 , DOI: 10.1021/acs.jctc.2c00924 Marc Reimann 1 , Martin Kaupp 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2022-11-23 , DOI: 10.1021/acs.jctc.2c00924 Marc Reimann 1 , Martin Kaupp 1
Affiliation
|
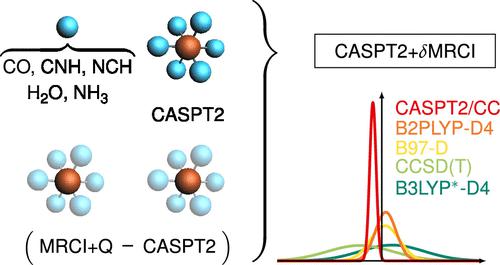
The CASPT2+δMRCI composite approach reported in a companion paper has been extended and used to provide high-quality reference data for a series of adiabatic spin gaps (defined as ΔE = Equintet – Esinglet) of [FeIIL6]2+ complexes (L = CNH, CO, NCH, NH3, H2O), either at nonrelativistic level or including scalar relativistic effects. These highly accurate data have been used to evaluate the performance of various more approximate methods. Coupled-cluster theory with singles, doubles, and perturbative triples, CCSD(T), is found to agree well with the new reference data for Werner-type complexes but exhibits larger underestimates by up to 70 kJ/mol for the π-acceptor ligands, due to appreciable static correlation in the low-spin states of these systems. Widely used domain-based local CCSD(T) calculations, DLPNO-CCSD(T), are shown to depend very sensitively on the cutoff values used to construct the localized domains, and standard values are not sufficient. A large number of density functional approximations have been evaluated against the new reference data. The B2PLYP double hybrid gives the smallest deviations, but several functionals from different rungs of the usual ladder hierarchy give mean absolute deviations below 20 kJ/mol. This includes the B97-D semilocal functional, the PBE0* global hybrid with 15% exact-exchange admixture, as well as the local hybrids LH07s-SVWN and LH07t-SVWN. Several further functionals achieve mean absolute errors below 30 kJ/mol (M06L-D4, SSB-D, B97-1-D4, LC-ωPBE-D4, LH12ct-SsirPW92-D4, LH12ct-SsifPW92-D4, LH14t-calPBE-D4, LHJ-HFcal-D4, and several further double hybrids) and thereby also still overall outperform CCSD(T) or uncorrected CASPT2. While exact-exchange admixture is a crucial factor in favoring high-spin states, the present evaluations confirm that other aspects can be important as well. A number of the better-performing functionals underestimate the spin gaps for the π-acceptor ligands but overestimate them for L = NH3, H2O. In contrast to a previous suggestion, non-self-consistent density functional theory (DFT) computations on top of Hartree–Fock orbitals are not a promising path to produce accurate spin gaps in such complexes.
中文翻译:

重新审视 3d 过渡金属配合物中的自旋态分裂:Fe(II) 配合物中绝热自旋态能量差异的基准近似方法
配套论文中报告的 CASPT2+δMRCI 复合方法已得到扩展,并用于为[Fe II L 6 ] 2的一系列绝热自旋间隙(定义为 Δ E = E五重态– E单重态)提供高质量参考数据+络合物 (L = CNH, CO, NCH, NH 3 , H 2O),要么在非相对论层面,要么包括标量相对论效应。这些高度准确的数据已被用于评估各种更近似方法的性能。发现具有单、双和微扰三元组的耦合簇理论 CCSD(T) 与 Werner 型复合物的新参考数据非常吻合,但对 π-受体配体表现出高达 70 kJ/mol 的更大低估,由于这些系统的低自旋状态下明显的静态相关性。广泛使用的基于域的局部 CCSD(T) 计算 DLPNO-CCSD(T) 显示非常敏感地依赖于用于构建局部域的截止值,而标准值是不够的。已经根据新的参考数据评估了大量密度泛函近似值。B2PLYP 双杂化给出了最小的偏差,但是来自通常阶梯层次的不同梯级的几个功能给出了低于 20 kJ/mol 的平均绝对偏差。这包括 B97-D 半局部功能、具有 15% 精确交换混合物的 PBE0* 全局混合,以及局部混合 LH07s-SVWN 和 LH07t-SVWN。其他几个泛函的平均绝对误差低于 30 kJ/mol(M06L-D4、SSB-D、B97-1-D4、LC-ωPBE-D4、LH12ct-SsirPW92-D4、LH12ct-SsifPW92-D4、LH14t-calPBE-D4 、LHJ-HFcal-D4 和其他几个双杂交),因此总体上仍然优于 CCSD(T) 或未校正的 CASPT2。虽然精确交换混合物是有利于高自旋状态的关键因素,但目前的评估证实其他方面也很重要。3 , H 2 O。与之前的建议相反,Hartree–Fock 轨道之上的非自洽密度泛函理论 (DFT) 计算并不是在此类复合物中产生准确自旋间隙的有前途的途径。
更新日期:2022-11-23
中文翻译:
重新审视 3d 过渡金属配合物中的自旋态分裂:Fe(II) 配合物中绝热自旋态能量差异的基准近似方法
配套论文中报告的 CASPT2+δMRCI 复合方法已得到扩展,并用于为[Fe II L 6 ] 2的一系列绝热自旋间隙(定义为 Δ E = E五重态– E单重态)提供高质量参考数据+络合物 (L = CNH, CO, NCH, NH 3 , H 2O),要么在非相对论层面,要么包括标量相对论效应。这些高度准确的数据已被用于评估各种更近似方法的性能。发现具有单、双和微扰三元组的耦合簇理论 CCSD(T) 与 Werner 型复合物的新参考数据非常吻合,但对 π-受体配体表现出高达 70 kJ/mol 的更大低估,由于这些系统的低自旋状态下明显的静态相关性。广泛使用的基于域的局部 CCSD(T) 计算 DLPNO-CCSD(T) 显示非常敏感地依赖于用于构建局部域的截止值,而标准值是不够的。已经根据新的参考数据评估了大量密度泛函近似值。B2PLYP 双杂化给出了最小的偏差,但是来自通常阶梯层次的不同梯级的几个功能给出了低于 20 kJ/mol 的平均绝对偏差。这包括 B97-D 半局部功能、具有 15% 精确交换混合物的 PBE0* 全局混合,以及局部混合 LH07s-SVWN 和 LH07t-SVWN。其他几个泛函的平均绝对误差低于 30 kJ/mol(M06L-D4、SSB-D、B97-1-D4、LC-ωPBE-D4、LH12ct-SsirPW92-D4、LH12ct-SsifPW92-D4、LH14t-calPBE-D4 、LHJ-HFcal-D4 和其他几个双杂交),因此总体上仍然优于 CCSD(T) 或未校正的 CASPT2。虽然精确交换混合物是有利于高自旋状态的关键因素,但目前的评估证实其他方面也很重要。3 , H 2 O。与之前的建议相反,Hartree–Fock 轨道之上的非自洽密度泛函理论 (DFT) 计算并不是在此类复合物中产生准确自旋间隙的有前途的途径。










































 京公网安备 11010802027423号
京公网安备 11010802027423号