当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Temperature Extrapolation of Molecular Dynamics Simulations of Complex Chemistry to Microsecond Timescales Using Kinetic Models: Applications to Hydrocarbon Pyrolysis
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2022-11-18 , DOI: 10.1021/acs.jctc.2c00623 Vincent Dufour-Décieux 1 , Brandi Ransom 1 , Austin D Sendek 1, 2 , Rodrigo Freitas 3 , Jose Blanchet 4 , Evan J Reed 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2022-11-18 , DOI: 10.1021/acs.jctc.2c00623 Vincent Dufour-Décieux 1 , Brandi Ransom 1 , Austin D Sendek 1, 2 , Rodrigo Freitas 3 , Jose Blanchet 4 , Evan J Reed 1
Affiliation
|
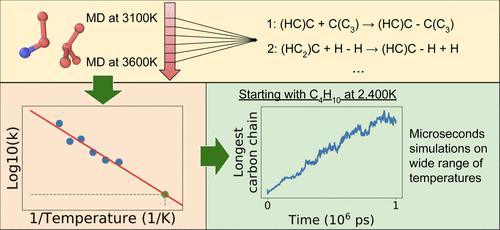
We develop a method to construct temperature-dependent kinetic models of hydrocarbon pyrolysis, based on information from molecular dynamics (MD) simulations of pyrolyzing systems in the high-temperature regime. MD simulations are currently a key tool to understand the mechanism of complex chemical processes such as pyrolysis and to observe their outcomes in different conditions, but these simulations are computationally expensive and typically limited to nanoseconds of simulation time. This limitation is inconsequential at high temperatures, where equilibrium is reached quickly, but at low temperatures, the system may not equilibrate within a tractable simulation timescale. In this work, we develop a method to construct kinetic models of hydrocarbon pyrolysis using the information from the high-temperature high-reactivity regime. We then extrapolate this model to low temperatures, which enables microsecond-long simulations to be performed. We show that this approach accurately predicts the time evolution of small molecules, as well as the size and composition of long carbon chains across a wide range of temperatures and compositions. Further, we show that the range of suitable temperatures for extrapolation can easily be improved by adding more simulations to the training data. Compared to experimental results, our kinetic model leads to similar compositional trends while allowing for more detailed kinetic and mechanistic insights.
中文翻译:

使用动力学模型将复杂化学的分子动力学模拟温度外推到微秒时间尺度:在烃热解中的应用
我们基于高温状态下热解系统的分子动力学 (MD) 模拟信息,开发了一种构建烃热解温度依赖性动力学模型的方法。MD 模拟目前是了解热解等复杂化学过程的机制并观察其在不同条件下的结果的关键工具,但这些模拟在计算上非常昂贵,而且通常仅限于纳秒级的模拟时间。这种限制在高温下无关紧要,在高温下会很快达到平衡,但在低温下,系统可能无法在易于处理的模拟时间尺度内达到平衡。在这项工作中,我们开发了一种方法,利用来自高温高反应性机制的信息构建碳氢化合物热解动力学模型。然后我们将该模型外推到低温,这使得能够执行微秒长的模拟。我们表明,这种方法准确地预测了小分子的时间演变,以及在广泛的温度和组成范围内长碳链的大小和组成。此外,我们表明,通过向训练数据添加更多模拟,可以轻松改进外推的合适温度范围。与实验结果相比,我们的动力学模型导致了类似的组成趋势,同时允许更详细的动力学和机械见解。以及在广泛的温度和组成范围内长碳链的大小和组成。此外,我们表明,通过向训练数据添加更多模拟,可以轻松改进外推的合适温度范围。与实验结果相比,我们的动力学模型导致了类似的组成趋势,同时允许更详细的动力学和机械见解。以及在广泛的温度和组成范围内长碳链的大小和组成。此外,我们表明,通过向训练数据添加更多模拟,可以轻松改进外推的合适温度范围。与实验结果相比,我们的动力学模型导致了类似的组成趋势,同时允许更详细的动力学和机械见解。
更新日期:2022-11-18
中文翻译:
使用动力学模型将复杂化学的分子动力学模拟温度外推到微秒时间尺度:在烃热解中的应用
我们基于高温状态下热解系统的分子动力学 (MD) 模拟信息,开发了一种构建烃热解温度依赖性动力学模型的方法。MD 模拟目前是了解热解等复杂化学过程的机制并观察其在不同条件下的结果的关键工具,但这些模拟在计算上非常昂贵,而且通常仅限于纳秒级的模拟时间。这种限制在高温下无关紧要,在高温下会很快达到平衡,但在低温下,系统可能无法在易于处理的模拟时间尺度内达到平衡。在这项工作中,我们开发了一种方法,利用来自高温高反应性机制的信息构建碳氢化合物热解动力学模型。然后我们将该模型外推到低温,这使得能够执行微秒长的模拟。我们表明,这种方法准确地预测了小分子的时间演变,以及在广泛的温度和组成范围内长碳链的大小和组成。此外,我们表明,通过向训练数据添加更多模拟,可以轻松改进外推的合适温度范围。与实验结果相比,我们的动力学模型导致了类似的组成趋势,同时允许更详细的动力学和机械见解。以及在广泛的温度和组成范围内长碳链的大小和组成。此外,我们表明,通过向训练数据添加更多模拟,可以轻松改进外推的合适温度范围。与实验结果相比,我们的动力学模型导致了类似的组成趋势,同时允许更详细的动力学和机械见解。以及在广泛的温度和组成范围内长碳链的大小和组成。此外,我们表明,通过向训练数据添加更多模拟,可以轻松改进外推的合适温度范围。与实验结果相比,我们的动力学模型导致了类似的组成趋势,同时允许更详细的动力学和机械见解。










































 京公网安备 11010802027423号
京公网安备 11010802027423号