当前位置:
X-MOL 学术
›
J. Mater. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Theoretical exploration of molecular packing and the charge transfer mechanism of organic solar cells based on PM6:Y6
Journal of Materials Chemistry A ( IF 11.9 ) Pub Date : 2022-11-14 , DOI: 10.1039/d2ta07420f Chongchen Xiang 1 , Qiming Zhao 1 , Wanqiang Liu 1 , Jiamin Cao 1 , Yingping Zou 2 , Hu Zhou 1
Journal of Materials Chemistry A ( IF 11.9 ) Pub Date : 2022-11-14 , DOI: 10.1039/d2ta07420f Chongchen Xiang 1 , Qiming Zhao 1 , Wanqiang Liu 1 , Jiamin Cao 1 , Yingping Zou 2 , Hu Zhou 1
Affiliation
|
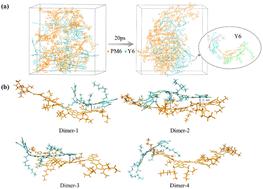
The active layer morphology of non-fullerene organic solar cells is one of the key factors affecting the power conversion efficiency (PCE); however, current experimental techniques cannot be used to directly observe the structural information at the electronic level. Molecular dynamics simulations and quantum chemical calculations provide effective means to explore the morphology and properties of active layers. In this paper, the local molecular stacking of PM6:Y6 films is simulated based on ab initio molecular dynamics (AIMD), and the simulation results show that the donor–acceptor (D–A) molecules are pi–pi stacked and some Y6 molecules are arranged in order. The excited state information of PM6:Y6 dimers was calculated by time-dependent density functional theory (TD-DFT) calculations. The results showed that ΔES1–CT < 0.1 eV, and dimers have very low exciton binding energy (Eb). The charge transfer processes of the D–A dimer are LE → CTX → CS and LE → CTX → CT1 → CS combined with hole–electron analysis. Moreover, ultraviolet-visible (UV-vis) spectra of J-type stacked dimers is similar to that of PM6:Y6 films. Finally, the electron transfer rates (kelectron) and hole transfer rates (khole) were calculated by Marcus theory, and the results showed that the PM6:Y6 system has high charge transfer rates, but the effect of molecular configuration on kelectron is less than that on khole. The properties of PM6:Y6 films were systematically investigated at the theoretical level in this work, and it demonstrated that PM6:Y6 films have pi–pi stacking, low ΔES1–CTX, low Eb, and high charge transfer rates.
中文翻译:

基于PM6:Y6的有机太阳能电池分子堆积与电荷转移机制的理论探索
非富勒烯有机太阳能电池的活性层形貌是影响功率转换效率(PCE)的关键因素之一;然而,目前的实验技术不能用于直接观察电子水平的结构信息。分子动力学模拟和量子化学计算为探索活性层的形貌和性质提供了有效手段。本文基于从头算模拟PM6:Y6薄膜的局部分子堆积分子动力学(AIMD),模拟结果表明供体-受体(D-A)分子呈pi-pi堆叠,一些Y6分子有序排列。PM6:Y6 二聚体的激发态信息是通过时间相关密度泛函理论 (TD-DFT) 计算得出的。结果表明,Δ E S 1 –CT < 0.1 eV,二聚体具有非常低的激子结合能(E b)。D-A 二聚体的电荷转移过程为 LE → CT X → CS 和 LE → CT X → CT 1 → CS 结合空穴-电子分析。此外,J 型堆叠二聚体的紫外-可见 (UV-vis) 光谱与 PM6:Y6 薄膜相似。最后,电子转移率(利用Marcus理论计算了k electron )和空穴转移率( k hole ),结果表明PM6:Y6体系具有较高的电荷转移率,但分子构型对k electron的影响小于对k hole的影响。本文在理论层面系统地研究了 PM6:Y6 薄膜的性质,证明 PM6:Y6 薄膜具有 pi-pi 堆叠、低 Δ E S 1 – CT X、低E b和高电荷转移率.
更新日期:2022-11-14
中文翻译:
基于PM6:Y6的有机太阳能电池分子堆积与电荷转移机制的理论探索
非富勒烯有机太阳能电池的活性层形貌是影响功率转换效率(PCE)的关键因素之一;然而,目前的实验技术不能用于直接观察电子水平的结构信息。分子动力学模拟和量子化学计算为探索活性层的形貌和性质提供了有效手段。本文基于从头算模拟PM6:Y6薄膜的局部分子堆积分子动力学(AIMD),模拟结果表明供体-受体(D-A)分子呈pi-pi堆叠,一些Y6分子有序排列。PM6:Y6 二聚体的激发态信息是通过时间相关密度泛函理论 (TD-DFT) 计算得出的。结果表明,Δ E S 1 –CT < 0.1 eV,二聚体具有非常低的激子结合能(E b)。D-A 二聚体的电荷转移过程为 LE → CT X → CS 和 LE → CT X → CT 1 → CS 结合空穴-电子分析。此外,J 型堆叠二聚体的紫外-可见 (UV-vis) 光谱与 PM6:Y6 薄膜相似。最后,电子转移率(利用Marcus理论计算了k electron )和空穴转移率( k hole ),结果表明PM6:Y6体系具有较高的电荷转移率,但分子构型对k electron的影响小于对k hole的影响。本文在理论层面系统地研究了 PM6:Y6 薄膜的性质,证明 PM6:Y6 薄膜具有 pi-pi 堆叠、低 Δ E S 1 – CT X、低E b和高电荷转移率.


























 京公网安备 11010802027423号
京公网安备 11010802027423号