当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Holo Protein Conformation Generation from Apo Structures by Ligand Binding Site Refinement
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2022-11-07 , DOI: 10.1021/acs.jcim.2c00895 Jinze Zhang 1 , Hao Li 1 , Xuejun Zhao 1 , Qilong Wu 1 , Sheng-You Huang 1
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2022-11-07 , DOI: 10.1021/acs.jcim.2c00895 Jinze Zhang 1 , Hao Li 1 , Xuejun Zhao 1 , Qilong Wu 1 , Sheng-You Huang 1
Affiliation
|
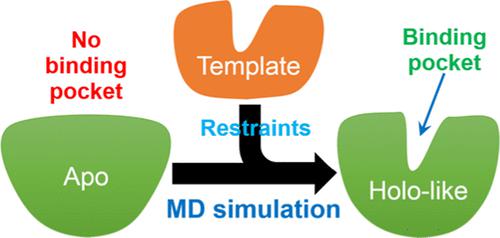
An important part in structure-based drug design is the selection of an appropriate protein structure. It has been revealed that a holo protein structure that contains a well-defined binding site is a much better choice than an apo structure in structure-based drug discovery. Therefore, it is valuable to obtain a holo-like protein conformation from apo structures in the case where no holo structure is available. Meeting the need, we present a robust approach to generate reliable holo-like structures from apo structures by ligand binding site refinement with restraints derived from holo templates with low homology. Our method was tested on a test set of 32 proteins from the DUD-E data set and compared with other approaches. It was shown that our method successfully refined the apo structures toward the corresponding holo conformations for 23 of 32 proteins, reducing the average all-heavy-atom RMSD of binding site residues by 0.48 Å. In addition, when evaluated against all the holo structures in the protein data bank, our method can improve the binding site RMSD for 14 of 19 cases that experience significant conformational changes. Furthermore, our refined structures also demonstrate their advantages over the apo structures in ligand binding mode predictions by both rigid docking and flexible docking and in virtual screening on the database of active and decoy ligands from the DUD-E. These results indicate that our method is effective in recovering holo-like conformations and will be valuable in structure-based drug discovery.
中文翻译:

通过配体结合位点细化从 Apo 结构生成全息蛋白构象
基于结构的药物设计的一个重要部分是选择合适的蛋白质结构。据透露,在基于结构的药物发现中,包含明确定义的结合位点的全蛋白结构比 apo 结构是更好的选择。因此,在没有全息结构可用的情况下,从 apo 结构获得类全息蛋白质构象是有价值的。为满足这一需要,我们提出了一种可靠的方法,通过配体结合位点细化以及源自低同源性全息模板的限制,从 apo 结构生成可靠的全息样结构。我们的方法在来自 DUD-E 数据集的 32 种蛋白质的测试集上进行了测试,并与其他方法进行了比较。结果表明,我们的方法成功地将 apo 结构细化为 32 种蛋白质中的 23 种的相应全息构象,将结合位点残基的平均全重原子 RMSD 降低了 0.48 Å。此外,当针对蛋白质数据库中的所有全息结构进行评估时,我们的方法可以改善 19 个经历显着构象变化的案例中的 14 个的结合位点 RMSD。此外,我们的精制结构还通过刚性对接和柔性对接以及在 DUD-E 的活性和诱饵配体数据库中进行虚拟筛选,在配体结合模式预测中展示了它们优于 apo 结构的优势。这些结果表明,我们的方法可有效恢复全息样构象,并将在基于结构的药物发现中发挥重要作用。将结合位点残基的平均全重原子 RMSD 降低 0.48 Å。此外,当针对蛋白质数据库中的所有全息结构进行评估时,我们的方法可以改善 19 个经历显着构象变化的案例中的 14 个的结合位点 RMSD。此外,我们的精制结构还通过刚性对接和柔性对接以及在 DUD-E 的活性和诱饵配体数据库中进行虚拟筛选,在配体结合模式预测中展示了它们优于 apo 结构的优势。这些结果表明,我们的方法可有效恢复全息样构象,并将在基于结构的药物发现中发挥重要作用。将结合位点残基的平均全重原子 RMSD 降低 0.48 Å。此外,当针对蛋白质数据库中的所有全息结构进行评估时,我们的方法可以改善 19 个经历显着构象变化的案例中的 14 个的结合位点 RMSD。此外,我们的精制结构还通过刚性对接和柔性对接以及在 DUD-E 的活性和诱饵配体数据库中进行虚拟筛选,在配体结合模式预测中展示了它们优于 apo 结构的优势。这些结果表明,我们的方法可有效恢复全息样构象,并将在基于结构的药物发现中发挥重要作用。我们的方法可以改善 19 个经历显着构象变化的案例中的 14 个的结合位点 RMSD。此外,我们的精制结构还通过刚性对接和柔性对接以及在 DUD-E 的活性和诱饵配体数据库中进行虚拟筛选,在配体结合模式预测中展示了它们优于 apo 结构的优势。这些结果表明,我们的方法可有效恢复全息样构象,并将在基于结构的药物发现中发挥重要作用。我们的方法可以改善 19 个经历显着构象变化的案例中的 14 个的结合位点 RMSD。此外,我们的精制结构还通过刚性对接和柔性对接以及在 DUD-E 的活性和诱饵配体数据库中进行虚拟筛选,在配体结合模式预测中展示了它们优于 apo 结构的优势。这些结果表明,我们的方法可有效恢复全息样构象,并将在基于结构的药物发现中发挥重要作用。
更新日期:2022-11-07
中文翻译:
通过配体结合位点细化从 Apo 结构生成全息蛋白构象
基于结构的药物设计的一个重要部分是选择合适的蛋白质结构。据透露,在基于结构的药物发现中,包含明确定义的结合位点的全蛋白结构比 apo 结构是更好的选择。因此,在没有全息结构可用的情况下,从 apo 结构获得类全息蛋白质构象是有价值的。为满足这一需要,我们提出了一种可靠的方法,通过配体结合位点细化以及源自低同源性全息模板的限制,从 apo 结构生成可靠的全息样结构。我们的方法在来自 DUD-E 数据集的 32 种蛋白质的测试集上进行了测试,并与其他方法进行了比较。结果表明,我们的方法成功地将 apo 结构细化为 32 种蛋白质中的 23 种的相应全息构象,将结合位点残基的平均全重原子 RMSD 降低了 0.48 Å。此外,当针对蛋白质数据库中的所有全息结构进行评估时,我们的方法可以改善 19 个经历显着构象变化的案例中的 14 个的结合位点 RMSD。此外,我们的精制结构还通过刚性对接和柔性对接以及在 DUD-E 的活性和诱饵配体数据库中进行虚拟筛选,在配体结合模式预测中展示了它们优于 apo 结构的优势。这些结果表明,我们的方法可有效恢复全息样构象,并将在基于结构的药物发现中发挥重要作用。将结合位点残基的平均全重原子 RMSD 降低 0.48 Å。此外,当针对蛋白质数据库中的所有全息结构进行评估时,我们的方法可以改善 19 个经历显着构象变化的案例中的 14 个的结合位点 RMSD。此外,我们的精制结构还通过刚性对接和柔性对接以及在 DUD-E 的活性和诱饵配体数据库中进行虚拟筛选,在配体结合模式预测中展示了它们优于 apo 结构的优势。这些结果表明,我们的方法可有效恢复全息样构象,并将在基于结构的药物发现中发挥重要作用。将结合位点残基的平均全重原子 RMSD 降低 0.48 Å。此外,当针对蛋白质数据库中的所有全息结构进行评估时,我们的方法可以改善 19 个经历显着构象变化的案例中的 14 个的结合位点 RMSD。此外,我们的精制结构还通过刚性对接和柔性对接以及在 DUD-E 的活性和诱饵配体数据库中进行虚拟筛选,在配体结合模式预测中展示了它们优于 apo 结构的优势。这些结果表明,我们的方法可有效恢复全息样构象,并将在基于结构的药物发现中发挥重要作用。我们的方法可以改善 19 个经历显着构象变化的案例中的 14 个的结合位点 RMSD。此外,我们的精制结构还通过刚性对接和柔性对接以及在 DUD-E 的活性和诱饵配体数据库中进行虚拟筛选,在配体结合模式预测中展示了它们优于 apo 结构的优势。这些结果表明,我们的方法可有效恢复全息样构象,并将在基于结构的药物发现中发挥重要作用。我们的方法可以改善 19 个经历显着构象变化的案例中的 14 个的结合位点 RMSD。此外,我们的精制结构还通过刚性对接和柔性对接以及在 DUD-E 的活性和诱饵配体数据库中进行虚拟筛选,在配体结合模式预测中展示了它们优于 apo 结构的优势。这些结果表明,我们的方法可有效恢复全息样构象,并将在基于结构的药物发现中发挥重要作用。










































 京公网安备 11010802027423号
京公网安备 11010802027423号