当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Redox-active ligands as a challenge for electronic structure methods
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2022-11-03 , DOI: 10.1002/jcc.27013 Ursula Rastetter 1 , Axel Jacobi von Wangelin 1 , Carmen Herrmann 1
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2022-11-03 , DOI: 10.1002/jcc.27013 Ursula Rastetter 1 , Axel Jacobi von Wangelin 1 , Carmen Herrmann 1
Affiliation
|
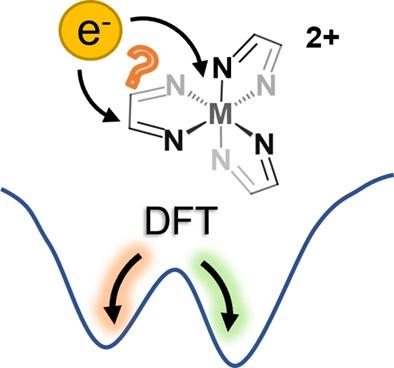
To improve the catalytic activity of 3d transition metal catalysts, redox-active ligands are a promising tool. These ligands influence the oxidation state of the metal center as well as the ground spin-state and make the experimental determination of both properties challenging. Therefore, first-principles calculations, in particular employing density functional theory with a proper choice of exchange-correlation (xc) functional, are crucial. Common xc functionals were tested on a simple class of metal complexes: homoleptic, octahedral tris(diimine) iron(II) complexes. The spin-state energy splittings for most of these complexes showed the expected linear dependence on the amount of exact exchange included in the xc functionals. Even though varying redox-activity affects the electronic structure of the complexes considerably, the sensitivity of the spin-state energetics to the exact exchange admixture is surprisingly small. For iron(II) complexes with highly redox-active ligands and for a broad range of ligands in the reduced tris(diimine) iron(I) complexes, self-consistent field convergence to local minima was observed, which differ from the global minimum in the redox state of the ligand. This may also result in convergence to a molecular structure that corresponds to an energetically higher-lying local minimum. One criterion to detect such behavior is a change in the sign of the slope for the dependence of the spin-state energy splittings on the amount of exact exchange. We discuss possible protocols for dealing with such artifacts in cases in which a large number of calculations makes checking by hand unfeasible.
中文翻译:

氧化还原活性配体作为电子结构方法的挑战
提高3d的催化活性过渡金属催化剂、氧化还原活性配体是一种很有前途的工具。这些配体会影响金属中心的氧化态以及基态自旋态,并使这两种性质的实验测定具有挑战性。因此,第一原理计算,特别是使用密度函数理论,具有适当的交换相关(XC)功能,至关重要。常见的 xc 泛函在一类简单的金属络合物上进行了测试:同配八面体三(二亚胺)铁(II)络合物。大多数这些复合物的自旋态能量分裂显示出对 xc 泛函中包含的精确交换量的预期线性依赖性。尽管不同的氧化还原活性会显着影响配合物的电子结构,自旋态能量学对精确交换混合物的敏感性出奇地小。对于具有高氧化还原活性配体的铁 (II) 配合物和还原的三 (二亚胺) 铁 (I) 配合物中的广泛配体,观察到自洽场收敛到局部最小值,这与全局最小值不同配体的氧化还原态。这也可能导致收敛到对应于能量较高的局部最小值的分子结构。检测这种行为的一个标准是自旋态能量分裂依赖于精确交换量的斜率符号的变化。我们讨论了在大量计算使得手动检查不可行的情况下处理此类工件的可能协议。对于具有高氧化还原活性配体的铁 (II) 配合物和还原的三 (二亚胺) 铁 (I) 配合物中的广泛配体,观察到自洽场收敛到局部最小值,这与全局最小值不同配体的氧化还原态。这也可能导致收敛到对应于能量较高的局部最小值的分子结构。检测这种行为的一个标准是自旋态能量分裂依赖于精确交换量的斜率符号的变化。我们讨论了在大量计算使得手动检查不可行的情况下处理此类工件的可能协议。对于具有高氧化还原活性配体的铁 (II) 配合物和还原的三 (二亚胺) 铁 (I) 配合物中的广泛配体,观察到自洽场收敛到局部最小值,这与全局最小值不同配体的氧化还原态。这也可能导致收敛到对应于能量较高的局部最小值的分子结构。检测这种行为的一个标准是自旋态能量分裂依赖于精确交换量的斜率符号的变化。我们讨论了在大量计算使得手动检查不可行的情况下处理此类工件的可能协议。观察到自洽场收敛到局部最小值,这与配体氧化还原状态下的全局最小值不同。这也可能导致收敛到对应于能量较高的局部最小值的分子结构。检测这种行为的一个标准是自旋态能量分裂依赖于精确交换量的斜率符号的变化。我们讨论了在大量计算使得手动检查不可行的情况下处理此类工件的可能协议。观察到自洽场收敛到局部最小值,这与配体氧化还原状态下的全局最小值不同。这也可能导致收敛到对应于能量较高的局部最小值的分子结构。检测这种行为的一个标准是自旋态能量分裂依赖于精确交换量的斜率符号的变化。我们讨论了在大量计算使得手动检查不可行的情况下处理此类工件的可能协议。检测这种行为的一个标准是自旋态能量分裂依赖于精确交换量的斜率符号的变化。我们讨论了在大量计算使得手动检查不可行的情况下处理此类工件的可能协议。检测这种行为的一个标准是自旋态能量分裂依赖于精确交换量的斜率符号的变化。我们讨论了在大量计算使得手动检查不可行的情况下处理此类工件的可能协议。
更新日期:2022-11-03
中文翻译:
氧化还原活性配体作为电子结构方法的挑战
提高3d的催化活性过渡金属催化剂、氧化还原活性配体是一种很有前途的工具。这些配体会影响金属中心的氧化态以及基态自旋态,并使这两种性质的实验测定具有挑战性。因此,第一原理计算,特别是使用密度函数理论,具有适当的交换相关(XC)功能,至关重要。常见的 xc 泛函在一类简单的金属络合物上进行了测试:同配八面体三(二亚胺)铁(II)络合物。大多数这些复合物的自旋态能量分裂显示出对 xc 泛函中包含的精确交换量的预期线性依赖性。尽管不同的氧化还原活性会显着影响配合物的电子结构,自旋态能量学对精确交换混合物的敏感性出奇地小。对于具有高氧化还原活性配体的铁 (II) 配合物和还原的三 (二亚胺) 铁 (I) 配合物中的广泛配体,观察到自洽场收敛到局部最小值,这与全局最小值不同配体的氧化还原态。这也可能导致收敛到对应于能量较高的局部最小值的分子结构。检测这种行为的一个标准是自旋态能量分裂依赖于精确交换量的斜率符号的变化。我们讨论了在大量计算使得手动检查不可行的情况下处理此类工件的可能协议。对于具有高氧化还原活性配体的铁 (II) 配合物和还原的三 (二亚胺) 铁 (I) 配合物中的广泛配体,观察到自洽场收敛到局部最小值,这与全局最小值不同配体的氧化还原态。这也可能导致收敛到对应于能量较高的局部最小值的分子结构。检测这种行为的一个标准是自旋态能量分裂依赖于精确交换量的斜率符号的变化。我们讨论了在大量计算使得手动检查不可行的情况下处理此类工件的可能协议。对于具有高氧化还原活性配体的铁 (II) 配合物和还原的三 (二亚胺) 铁 (I) 配合物中的广泛配体,观察到自洽场收敛到局部最小值,这与全局最小值不同配体的氧化还原态。这也可能导致收敛到对应于能量较高的局部最小值的分子结构。检测这种行为的一个标准是自旋态能量分裂依赖于精确交换量的斜率符号的变化。我们讨论了在大量计算使得手动检查不可行的情况下处理此类工件的可能协议。观察到自洽场收敛到局部最小值,这与配体氧化还原状态下的全局最小值不同。这也可能导致收敛到对应于能量较高的局部最小值的分子结构。检测这种行为的一个标准是自旋态能量分裂依赖于精确交换量的斜率符号的变化。我们讨论了在大量计算使得手动检查不可行的情况下处理此类工件的可能协议。观察到自洽场收敛到局部最小值,这与配体氧化还原状态下的全局最小值不同。这也可能导致收敛到对应于能量较高的局部最小值的分子结构。检测这种行为的一个标准是自旋态能量分裂依赖于精确交换量的斜率符号的变化。我们讨论了在大量计算使得手动检查不可行的情况下处理此类工件的可能协议。检测这种行为的一个标准是自旋态能量分裂依赖于精确交换量的斜率符号的变化。我们讨论了在大量计算使得手动检查不可行的情况下处理此类工件的可能协议。检测这种行为的一个标准是自旋态能量分裂依赖于精确交换量的斜率符号的变化。我们讨论了在大量计算使得手动检查不可行的情况下处理此类工件的可能协议。


























 京公网安备 11010802027423号
京公网安备 11010802027423号