当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
DyScore: A Boosting Scoring Method with Dynamic Properties for Identifying True Binders and Nonbinders in Structure-Based Drug Discovery
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2022-11-03 , DOI: 10.1021/acs.jcim.2c00926 Yanjun Li 1, 2 , Daohong Zhou 3 , Guangrong Zheng 4 , Xiaolin Li 5 , Dapeng Wu 1 , Yaxia Yuan 3
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2022-11-03 , DOI: 10.1021/acs.jcim.2c00926 Yanjun Li 1, 2 , Daohong Zhou 3 , Guangrong Zheng 4 , Xiaolin Li 5 , Dapeng Wu 1 , Yaxia Yuan 3
Affiliation
|
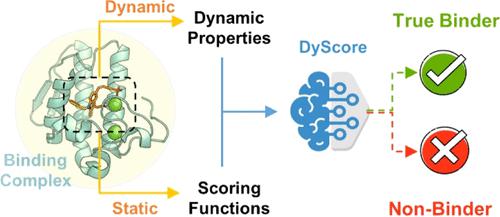
The accurate prediction of protein–ligand binding affinity is critical for the success of computer-aided drug discovery. However, the accuracy of current scoring functions is usually unsatisfactory due to their rough approximation or sometimes even omittance of many factors involved in protein–ligand binding. For instance, the intrinsic dynamics of the protein–ligand binding state is usually disregarded in scoring function because these rapid binding affinity prediction approaches are only based on a representative complex structure of the protein and ligand in the binding state. That is, the dynamic protein–ligand binding complex ensembles are simplified as a static snapshot in calculation. In this study, two novel features were proposed for characterizing the dynamic properties of protein–ligand binding based on the static structure of the complex, which is expected to be a valuable complement to the current scoring functions. The two features demonstrate the geometry-shape matching between a protein and a ligand as well as the dynamic stability of protein–ligand binding. We further combined these two novel features with several classical scoring functions to develop a binary classification model called DyScore that uses the Extreme Gradient Boosting algorithm to classify compound poses as binders or non-binders. We have found that DyScore achieves state-of-the-art performance in distinguishing active and decoy ligands on both enhanced DUD data set and external test sets with both proposed novel features showing significant contributions to the improved performance. Especially, DyScore exhibits superior performance on early recognition, a crucial requirement for success in virtual screening and de novo drug design. The standalone version of DyScore and Dyscore-MF are freely available to all at: https://github.com/YanjunLi-CS/dyscore.
中文翻译:

DyScore:一种具有动态特性的提升评分方法,用于在基于结构的药物发现中识别真正的结合剂和非结合剂
准确预测蛋白质-配体结合亲和力对于计算机辅助药物发现的成功至关重要。然而,当前评分函数的准确性通常不尽如人意,因为它们粗略近似,有时甚至忽略了与蛋白质-配体结合有关的许多因素。例如,蛋白质-配体结合状态的内在动力学通常在评分函数中被忽略,因为这些快速结合亲和力预测方法仅基于结合状态下蛋白质和配体的代表性复杂结构。也就是说,动态蛋白质-配体结合复合体在计算中被简化为静态快照。在这项研究中,基于复合物的静态结构,提出了两个新特征来表征蛋白质-配体结合的动态特性,这有望成为对当前评分函数的有价值的补充。这两个特征证明了蛋白质和配体之间的几何形状匹配以及蛋白质-配体结合的动态稳定性。我们进一步将这两个新特征与几个经典评分函数相结合,开发了一个名为 DyScore 的二元分类模型,该模型使用极端梯度提升算法将复合姿势分类为绑定或非绑定。我们发现 DyScore 在增强 DUD 数据集和外部测试集上区分活性配体和诱饵配体方面实现了最先进的性能,并且提出的两个新功能都显示出对改进性能的重大贡献。特别是,DyScore 在早期识别方面表现出卓越的性能,这是虚拟筛选成功的关键要求和从头药物设计。DyScore 和 Dyscore-MF 的独立版本可供所有人免费使用:https://github.com/YanjunLi-CS/dyscore。
更新日期:2022-11-03
中文翻译:
DyScore:一种具有动态特性的提升评分方法,用于在基于结构的药物发现中识别真正的结合剂和非结合剂
准确预测蛋白质-配体结合亲和力对于计算机辅助药物发现的成功至关重要。然而,当前评分函数的准确性通常不尽如人意,因为它们粗略近似,有时甚至忽略了与蛋白质-配体结合有关的许多因素。例如,蛋白质-配体结合状态的内在动力学通常在评分函数中被忽略,因为这些快速结合亲和力预测方法仅基于结合状态下蛋白质和配体的代表性复杂结构。也就是说,动态蛋白质-配体结合复合体在计算中被简化为静态快照。在这项研究中,基于复合物的静态结构,提出了两个新特征来表征蛋白质-配体结合的动态特性,这有望成为对当前评分函数的有价值的补充。这两个特征证明了蛋白质和配体之间的几何形状匹配以及蛋白质-配体结合的动态稳定性。我们进一步将这两个新特征与几个经典评分函数相结合,开发了一个名为 DyScore 的二元分类模型,该模型使用极端梯度提升算法将复合姿势分类为绑定或非绑定。我们发现 DyScore 在增强 DUD 数据集和外部测试集上区分活性配体和诱饵配体方面实现了最先进的性能,并且提出的两个新功能都显示出对改进性能的重大贡献。特别是,DyScore 在早期识别方面表现出卓越的性能,这是虚拟筛选成功的关键要求和从头药物设计。DyScore 和 Dyscore-MF 的独立版本可供所有人免费使用:https://github.com/YanjunLi-CS/dyscore。








































 京公网安备 11010802027423号
京公网安备 11010802027423号