Journal of Advanced Research ( IF 11.4 ) Pub Date : 2022-10-30 , DOI: 10.1016/j.jare.2022.10.012 Javier Cuenca-Guardiola 1 , Belén de la Morena-Barrio 2 , Juan L García 3 , Alba Sanchis-Juan 4 , Javier Corral 5 , Jesualdo T Fernández-Breis 1
|
Introduction
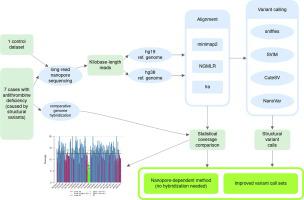
Whole-genome sequencing using nanopore technologies can uncover structural variants, which are DNA rearrangements larger than 50 base pairs. Nanopore technologies can also characterize their boundaries with single-base accuracy, owing to the kilobase-long reads that encompass either full variants or their junctions. Other methods, such as next-generation short read sequencing or PCR assays, are limited in their capabilities to detect or characterize structural variants. However, the existing software for nanopore sequencing data analysis still reports incomplete variant sets, which also contain erroneous calls, a considerable obstacle for the molecular diagnosis or accurate genotyping of populations.
Methods
We compared multiple factors affecting variant calling, such as reference genome version, aligner (minimap2, NGMLR, and lra) choice, and variant caller combinations (Sniffles, CuteSV, SVIM, and NanoVar), to find the optimal group of tools for calling large (>50 kb) deletions and duplications, using data from seven patients exhibiting gross gene defects on SERPINC1 and from a reference variant set as the control. The goal was to obtain the most complete, yet reasonably specific group of large variants using a single cell of PromethION sequencing, which yielded lower depth coverage than short-read sequencing. We also used a custom method for the statistical analysis of the coverage value to refine the resulting datasets.
Results
We found that for large deletions and duplications (>50 kb), the existing software performed worse than for smaller ones, in terms of both sensitivity and specificity, and newer tools had not improved this. Our novel software, disCoverage, could polish variant callers’ results, improving specificity by up to 62% and sensitivity by 15%, the latter requiring other data or samples.
Conclusion
We analyzed the current situation of >50-kb copy number variants with nanopore sequencing, which could be improved. The methods presented in this work could help to identify the known deletions and duplications in a set of patients, while also helping to filter out erroneous calls for these variants, which might aid the efforts to characterize a not-yet well-known fraction of genetic variability in the human genome.
中文翻译:
全基因组纳米孔测序改进大拷贝数变异检测
介绍
使用纳米孔技术的全基因组测序可以发现结构变异,即大于 50 个碱基对的 DNA 重排。纳米孔技术还可以以单碱基精度表征其边界,因为千碱基长的读数包含完整的变体或其连接点。其他方法,例如下一代短读长测序或 PCR 测定,检测或表征结构变异的能力有限。然而,现有的纳米孔测序数据分析软件仍然报告不完整的变异集,其中还包含错误的识别,这对群体的分子诊断或准确的基因分型来说是一个相当大的障碍。
方法
我们比较了影响变异检出的多种因素,例如参考基因组版本、对齐器(minimap2、NGMLR 和 lra)选择以及变异检出器组合(Sniffles、CuteSV、SVIM 和 NanoVar),以找到用于检出大样本的最佳工具组。 (>50 kb) 删除和重复,使用来自 7 名在SERPINC1上表现出严重基因缺陷的患者的数据以及来自作为对照的参考变体集的数据。目标是使用 PromethION 测序的单细胞获得最完整但相当特定的大变异组,该测序产生的深度覆盖率低于短读长测序。我们还使用自定义方法对覆盖值进行统计分析,以细化生成的数据集。
结果
我们发现,对于大的删除和重复(> 50 kb),现有软件在灵敏度和特异性方面都比较小的软件表现更差,而新的工具并没有改善这一点。我们的新颖软件 disCoverage 可以完善变异识别者的结果,将特异性提高高达 62%,将灵敏度提高 15%,后者需要其他数据或样本。
结论
我们通过纳米孔测序分析了>50-kb拷贝数变异的现状,并提出了可以改进的地方。这项工作中提出的方法可以帮助识别一组患者中已知的缺失和重复,同时还有助于过滤掉对这些变异的错误调用,这可能有助于描述尚未为人所知的遗传片段的特征。人类基因组的变异性。










































 京公网安备 11010802027423号
京公网安备 11010802027423号