当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
An automated method for graph-based chemical space exploration and transition state finding
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2022-10-14 , DOI: 10.1002/jcc.27011 Pablo Ramos-Sánchez 1, 2 , Jeremy N Harvey 2 , José A Gámez 1
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2022-10-14 , DOI: 10.1002/jcc.27011 Pablo Ramos-Sánchez 1, 2 , Jeremy N Harvey 2 , José A Gámez 1
Affiliation
|
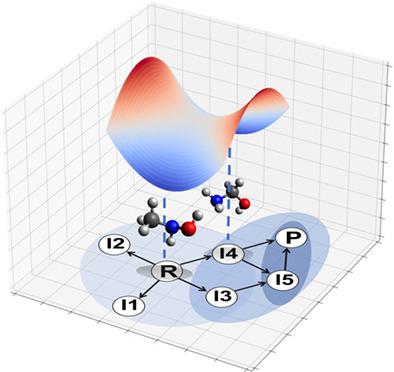
Algorithms that automatically explore the chemical space have been limited to chemical systems with a low number of atoms due to expensive involved quantum calculations and the large amount of possible reaction pathways. The method described here presents a novel solution to the problem of chemical exploration by generating reaction networks with heuristics based on chemical theory. First, a second version of the reaction network is determined through molecular graph transformations acting upon functional groups of the reacting. Only transformations that break two chemical bonds and form two new ones are considered, leading to a significant performance enhancement compared to previously presented algorithm. Second, energy barriers for this reaction network are estimated through quantum chemical calculations by a growing string method, which can also identify non-octet species missed during the previous step and further define the reaction network. The proposed algorithm has been successfully applied to five different chemical reactions, in all cases identifying the most important reaction pathways.
中文翻译:

基于图的化学空间探索和过渡态发现的自动化方法
由于昂贵的量子计算和大量可能的反应途径,自动探索化学空间的算法仅限于原子数较少的化学系统。此处描述的方法通过基于化学理论的启发式生成反应网络,为化学探索问题提供了一种新颖的解决方案。首先,反应网络的第二个版本是通过作用于反应官能团的分子图变换来确定的。仅考虑打破两个化学键并形成两个新化学键的转换,与之前提出的算法相比,性能显着提高。其次,该反应网络的能垒是通过增长弦法的量子化学计算来估计的,它还可以识别在上一步中遗漏的非八位组物种,并进一步定义反应网络。所提出的算法已成功应用于五种不同的化学反应,在所有情况下都确定了最重要的反应途径。
更新日期:2022-10-14
中文翻译:
基于图的化学空间探索和过渡态发现的自动化方法
由于昂贵的量子计算和大量可能的反应途径,自动探索化学空间的算法仅限于原子数较少的化学系统。此处描述的方法通过基于化学理论的启发式生成反应网络,为化学探索问题提供了一种新颖的解决方案。首先,反应网络的第二个版本是通过作用于反应官能团的分子图变换来确定的。仅考虑打破两个化学键并形成两个新化学键的转换,与之前提出的算法相比,性能显着提高。其次,该反应网络的能垒是通过增长弦法的量子化学计算来估计的,它还可以识别在上一步中遗漏的非八位组物种,并进一步定义反应网络。所提出的算法已成功应用于五种不同的化学反应,在所有情况下都确定了最重要的反应途径。










































 京公网安备 11010802027423号
京公网安备 11010802027423号