当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Benchmark of density functional theory methods for the study of organic polysulfides
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2022-09-28 , DOI: 10.1002/jcc.27007 Jyoti Sharma 1 , Pier Alexandre Champagne 1
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2022-09-28 , DOI: 10.1002/jcc.27007 Jyoti Sharma 1 , Pier Alexandre Champagne 1
Affiliation
|
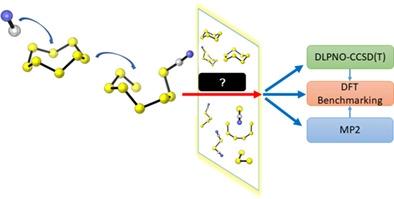
Elemental sulfur is often used in organic synthesis as its low cost and high abundance make it a highly desirable source of sulfur atoms. However, sulfur's unpredictable catenation behavior poses challenges to its widespread usage due to difficulties in designing new reactions that can account for its multifaceted reactivity. In order to accurately model sulfur's mechanisms using computational approaches, it is necessary to identify density functional theory (DFT) methods that are accurate on these systems. This study benchmarks 12 well-known DFT functionals that include local, non-local, and hybrid methods against DLPNO-CCSD(T)/aug-cc-pV(Q+d)Z//MP2/aug-cc-pV(T+d)Z/SMD(MeCN) for the accurate treatment of organic polysulfides, taking cyanide as a nucleophile. Our benchmarking results indicate that the M06-2X and B3LYP-D3(BJ) density functionals are the most accurate for calculating reaction energies, while local functionals performed the worst. For activation energies, MN15, MN15-L, M06-2X, and ωB97X-D are the most accurate. Our analysis of structural parameters shows that all functionals perform well for ground state optimizations except B97D3, while MN15-L and M06-2X performed best for transition structure optimizations. Overall, the four hybrid functionals MN15, M06-2X, ωB97X-D, and B3LYP-D3(BJ) appear adequate for studying the reaction mechanisms of polysulfides.
中文翻译:

有机多硫化物研究的密度泛函理论方法基准
元素硫经常用于有机合成,因为其低成本和高丰度使其成为非常理想的硫原子来源。然而,由于难以设计能够解释其多方面反应性的新反应,硫不可预测的连锁行为对其广泛使用提出了挑战。为了使用计算方法准确模拟硫的机制,有必要确定在这些系统上准确的密度泛函理论 (DFT) 方法。这项研究对 12 种著名的 DFT 泛函进行了基准测试,其中包括针对 DLPNO-CCSD(T)/aug-cc-pV(Q+d)Z//MP2/aug-cc-pV(T) 的局部、非局部和混合方法+d)Z/SMD(MeCN) 用于精确处理有机多硫化物,以氰化物为亲核试剂。我们的基准测试结果表明 M06-2X 和 B3LYP-D3(BJ) 密度泛函在计算反应能方面最准确,而局部泛函表现最差。对于活化能,MN15、MN15-L、M06-2X 和 ωB97X-D 是最准确的。我们对结构参数的分析表明,除 B97D3 外,所有泛函都在基态优化方面表现良好,而 MN15-L 和 M06-2X 在过渡结构优化方面表现最佳。总体而言,四种杂化官能团 MN15、M06-2X、ωB97X-D 和 B3LYP-D3(BJ) 似乎足以研究多硫化物的反应机理。我们对结构参数的分析表明,除 B97D3 外,所有泛函都在基态优化方面表现良好,而 MN15-L 和 M06-2X 在过渡结构优化方面表现最佳。总体而言,四种杂化官能团 MN15、M06-2X、ωB97X-D 和 B3LYP-D3(BJ) 似乎足以研究多硫化物的反应机理。我们对结构参数的分析表明,除 B97D3 外,所有泛函都在基态优化方面表现良好,而 MN15-L 和 M06-2X 在过渡结构优化方面表现最佳。总体而言,四种杂化官能团 MN15、M06-2X、ωB97X-D 和 B3LYP-D3(BJ) 似乎足以研究多硫化物的反应机理。
更新日期:2022-09-28
中文翻译:
有机多硫化物研究的密度泛函理论方法基准
元素硫经常用于有机合成,因为其低成本和高丰度使其成为非常理想的硫原子来源。然而,由于难以设计能够解释其多方面反应性的新反应,硫不可预测的连锁行为对其广泛使用提出了挑战。为了使用计算方法准确模拟硫的机制,有必要确定在这些系统上准确的密度泛函理论 (DFT) 方法。这项研究对 12 种著名的 DFT 泛函进行了基准测试,其中包括针对 DLPNO-CCSD(T)/aug-cc-pV(Q+d)Z//MP2/aug-cc-pV(T) 的局部、非局部和混合方法+d)Z/SMD(MeCN) 用于精确处理有机多硫化物,以氰化物为亲核试剂。我们的基准测试结果表明 M06-2X 和 B3LYP-D3(BJ) 密度泛函在计算反应能方面最准确,而局部泛函表现最差。对于活化能,MN15、MN15-L、M06-2X 和 ωB97X-D 是最准确的。我们对结构参数的分析表明,除 B97D3 外,所有泛函都在基态优化方面表现良好,而 MN15-L 和 M06-2X 在过渡结构优化方面表现最佳。总体而言,四种杂化官能团 MN15、M06-2X、ωB97X-D 和 B3LYP-D3(BJ) 似乎足以研究多硫化物的反应机理。我们对结构参数的分析表明,除 B97D3 外,所有泛函都在基态优化方面表现良好,而 MN15-L 和 M06-2X 在过渡结构优化方面表现最佳。总体而言,四种杂化官能团 MN15、M06-2X、ωB97X-D 和 B3LYP-D3(BJ) 似乎足以研究多硫化物的反应机理。我们对结构参数的分析表明,除 B97D3 外,所有泛函都在基态优化方面表现良好,而 MN15-L 和 M06-2X 在过渡结构优化方面表现最佳。总体而言,四种杂化官能团 MN15、M06-2X、ωB97X-D 和 B3LYP-D3(BJ) 似乎足以研究多硫化物的反应机理。








































 京公网安备 11010802027423号
京公网安备 11010802027423号