Solid State Nuclear Magnetic Resonance ( IF 1.8 ) Pub Date : 2022-09-24 , DOI: 10.1016/j.ssnmr.2022.101832 Joshua D Hartman 1 , James K Harper 2
|
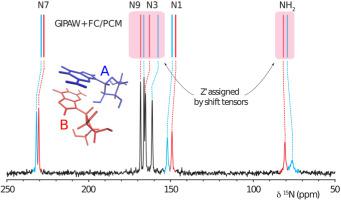
Ab initio methods for predicting NMR parameters in the solid state are an essential tool for assigning experimental spectra and play an increasingly important role in structural characterizations. Recently, a molecular correction (MC) technique has been developed which combines the strengths of plane-wave methods (GIPAW) with single molecule calculations employing Gaussian basis sets. The GIPAW + MC method relies on a periodic calculation performed at a lower level of theory to model the crystalline environment. The GIPAW result is then corrected using a single molecule calculation performed at a higher level of theory. The success of the GIPAW + MC method in predicting a range of NMR parameters is a result of the highly local character of the tensors underlying the NMR observable. However, in applications involving strong intermolecular interactions we find that expanding the region treated at the higher level of theory more accurately captures local many-body contributions to the NMR chemical shielding (CS) tensor. We propose alternative corrections to GIPAW which capture interactions between adjacent molecules at a higher level of theory using either fragment or cluster-based calculations. Benchmark calculations performed on and data sets show that these advanced GIPAW-corrected calculations improve the accuracy of chemical shielding tensor predictions relative to existing methods. Specifically, cluster-based corrections show a 24% and 17% reduction in RMS error relative to GIPAW and GIPAW + MC calculations, respectively. Comparing the benchmark data sets using multiple computational models demonstrates that CS tensor calculations are significantly more sensitive to intermolecular interactions relative to . However, fragment and cluster-based corrections that include direct hydrogen bond partners are sufficient for optimizing the accuracy of GIPAW-corrected methods. Finally, GIPAW-corrected methods are applied to the particularly challenging NMR spectral assignment of guanosine dihydrate which contains two guanosine molecules in the asymmetric unit.
中文翻译:
通过簇和碎片校正提高 GIPAW 化学屏蔽计算的准确性
用于预测固态 NMR 参数的从头算方法是分配实验光谱的重要工具,并在结构表征中发挥越来越重要的作用。最近,开发了一种分子校正 (MC) 技术,它将平面波方法 (GIPAW) 的优势与采用高斯基组的单分子计算相结合。GIPAW + MC 方法依赖于在较低理论水平上执行的周期性计算来模拟晶体环境。然后使用在更高理论水平上执行的单分子计算来校正 GIPAW 结果。GIPAW + MC 方法在预测一系列 NMR 参数方面的成功是基于 NMR 可观测张量的高度局部特征的结果。然而,NMR 化学屏蔽 (CS) 张量。我们提出了对 GIPAW 的替代修正,它使用基于片段或基于簇的计算在更高的理论水平上捕获相邻分子之间的相互作用。基准计算执行于和数据集表明,这些先进的 GIPAW 校正计算相对于现有方法提高了化学屏蔽张量预测的准确性。具体来说,基于集群相对于 GIPAW 和 GIPAW + MC 计算,校正显示 RMS 误差分别减少了 24% 和 17%。使用多个计算模型比较基准数据集表明CS 张量计算相对于分子间相互作用明显更敏感. 然而,包括直接氢键伙伴在内的基于片段和簇的校正足以优化 GIPAW 校正方法的准确性。最后,将 GIPAW 校正方法应用于特别具有挑战性的鸟苷二水合物的 NMR 光谱分配,该二水合物在不对称单元中包含两个鸟苷分子。









































 京公网安备 11010802027423号
京公网安备 11010802027423号