当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
A coarse-grained approach to NMR-data-assisted modeling of protein structures
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2022-09-22 , DOI: 10.1002/jcc.27003 Emilia A Lubecka 1 , Adam Liwo 2
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2022-09-22 , DOI: 10.1002/jcc.27003 Emilia A Lubecka 1 , Adam Liwo 2
Affiliation
|
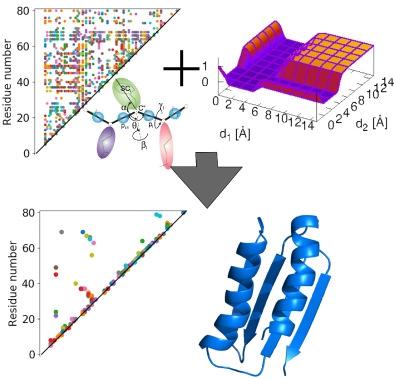
The ESCASA algorithm for analytical estimation of proton positions from coarse-grained geometry developed in our recent work has been implemented in modeling protein structures with the highly coarse-grained UNRES model of polypeptide chains (two sites per residue) and nuclear magnetic resonance (NMR) data. A penalty function with the shape of intersecting gorges was applied to treat ambiguous distance restraints, which automatically selects consistent restraints. Hamiltonian replica exchange molecular dynamics was used to carry out the conformational search. The method was tested with both unambiguous and ambiguous restraints producing good-quality models with GDT_TS from 7.4 units higher to 14.4 units lower than those obtained with the CYANA or MELD software for protein-structure determination from NMR data at the all-atom resolution. The method can thus be applied in modeling the structures of flexible proteins, for which extensive conformational search enabled by coarse-graining is more important than high modeling accuracy.
中文翻译:

核磁共振数据辅助蛋白质结构建模的粗粒度方法
在我们最近的工作中开发的用于从粗粒度几何分析估计质子位置的 ESCASA 算法已经在使用多肽链的高度粗粒度 UNRES 模型(每个残基两个位点)和核磁共振(NMR)的蛋白质结构建模中得到实施数据。采用相交峡谷形状的惩罚函数来处理模糊的距离约束,自动选择一致的约束。哈密顿副本交换分子动力学用于进行构象搜索。该方法在明确和模糊的约束下进行了测试,产生了 GDT_TS 的高质量模型,比使用 CYANA 或 MELD 软件获得的模型高 7.4 个单位,从全原子分辨率的 NMR 数据确定蛋白质结构。
更新日期:2022-09-22
中文翻译:
核磁共振数据辅助蛋白质结构建模的粗粒度方法
在我们最近的工作中开发的用于从粗粒度几何分析估计质子位置的 ESCASA 算法已经在使用多肽链的高度粗粒度 UNRES 模型(每个残基两个位点)和核磁共振(NMR)的蛋白质结构建模中得到实施数据。采用相交峡谷形状的惩罚函数来处理模糊的距离约束,自动选择一致的约束。哈密顿副本交换分子动力学用于进行构象搜索。该方法在明确和模糊的约束下进行了测试,产生了 GDT_TS 的高质量模型,比使用 CYANA 或 MELD 软件获得的模型高 7.4 个单位,从全原子分辨率的 NMR 数据确定蛋白质结构。










































 京公网安备 11010802027423号
京公网安备 11010802027423号