Clinical Pharmacokinetics ( IF 4.6 ) Pub Date : 2022-09-15 , DOI: 10.1007/s40262-022-01157-8 Mingxiang Liao 1 , Jeri Beltman 1 , Heidi Giordano 1 , Thomas C Harding 1 , Lara Maloney 1 , Andrew D Simmons 1 , Jim J Xiao 1
|
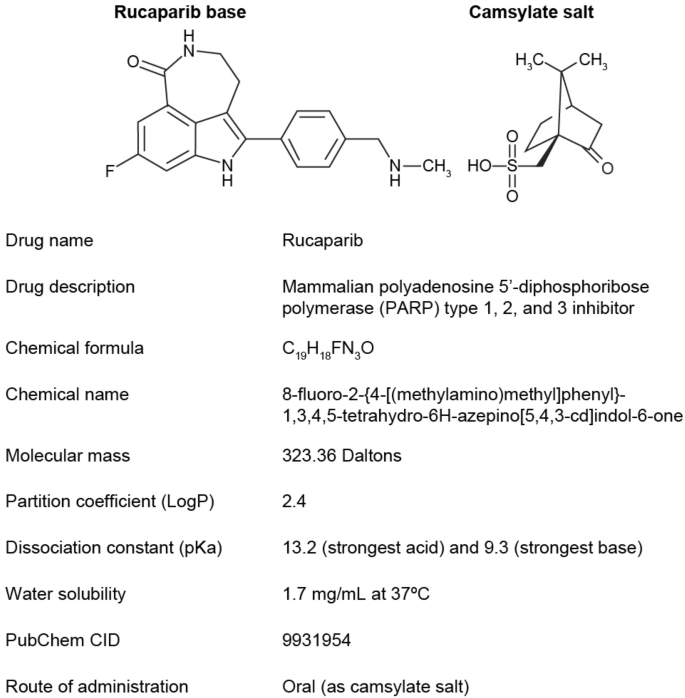
Rucaparib is an oral small-molecule poly(ADP-ribose) polymerase inhibitor indicated for patients with recurrent ovarian cancer in the maintenance and treatment settings and for patients with metastatic castration-resistant prostate cancer associated with a deleterious BRCA1 or BRCA2 mutation. Rucaparib has a manageable safety profile; the most common adverse events reported were fatigue and nausea in both indications. Accumulation in plasma exposure occurred after repeated administration of the approved 600-mg twice-daily dosage. Steady state was achieved after continuous twice-daily dosing for a week. Rucaparib has moderate oral bioavailability and can be dosed with or without food. Although a high-fat meal weakly increased maximum concentration and area under the curve, the effect was not clinically significant. A mass balance analysis indicated almost a complete dose recovery of rucaparib over 12 days, with metabolism, renal, and hepatic excretion as the elimination routes. A population pharmacokinetic analysis of rucaparib revealed no effect of age, sex, race, or body weight. No starting dose adjustments were necessary for patients with mild-to-moderate hepatic or renal impairment; the effect of severe organ impairment on rucaparib exposure has not been evaluated. In patients, rucaparib moderately inhibited cytochrome P450 (CYP) 1A2 and weakly inhibited CYP3As, CYP2C9, and CYP2C19. Rucaparib weakly increased systemic exposures of oral contraceptives and oral rosuvastatin and marginally increased the exposure of oral digoxin (a P-glycoprotein substrate). In vitro studies suggested that rucaparib inhibits transporters MATE1, MATE2-K, OCT1, and OCT2. No clinically meaningful drug interactions with rucaparib as a perpetrator were observed. An exposure–response analysis revealed dose-dependent changes in selected clinical efficacy and safety endpoints. Overall, this article provides a comprehensive review of the clinical pharmacokinetics, pharmacodynamics, drug–drug interactions, effects of intrinsic and extrinsic factors, and exposure–response relationships of rucaparib.
中文翻译:
Rucaparib的临床药代动力学和药效学
Rucaparib 是一种口服小分子聚(ADP-核糖)聚合酶抑制剂,适用于维持和治疗中的复发性卵巢癌患者以及与有害 BRCA1 或 BRCA2 突变相关的转移性去势抵抗性前列腺癌患者。Rucaparib 具有可控的安全性;报告的最常见不良事件是这两种适应症的疲劳和恶心。重复给予批准的 600 毫克每日两次剂量后,血浆暴露量发生累积。连续每日两次给药一周后达到稳定状态。Rucaparib 具有中等的口服生物利用度,可以与食物一起服用或单独服用。尽管高脂肪膳食微弱地增加了最大浓度和曲线下面积,但其效果在临床上并不显着。质量平衡分析表明rucaparib在12天内几乎完全剂量恢复,代谢、肾和肝排泄作为消除途径。rucaparib 的群体药代动力学分析显示年龄、性别、种族或体重没有影响。对于轻度至中度肝或肾功能不全的患者,无需调整起始剂量;尚未评估严重器官损伤对rucaparib暴露的影响。在患者中,rucaparib 中度抑制细胞色素 P450 (CYP) 1A2,并弱抑制 CYP3As、CYP2C9 和 CYP2C19。Rucaparib 微弱地增加了口服避孕药和口服瑞舒伐他汀的全身暴露,并略微增加了口服地高辛(一种 P-糖蛋白底物)的暴露。体外研究表明 rucaparib 抑制转运蛋白 MATE1、MATE2-K、OCT1 和 OCT2。没有观察到与 rucaparib 作为肇事者有临床意义的药物相互作用。暴露-反应分析揭示了选定的临床疗效和安全性终点的剂量依赖性变化。总体而言,本文对 rucaparib 的临床药代动力学、药效学、药物相互作用、内在和外在因素的影响以及暴露-反应关系进行了全面的综述。










































 京公网安备 11010802027423号
京公网安备 11010802027423号